manligt Turners syndrom, Noonan-Ehmke syndrom, Turner-liknande syndrom, Ullrich-Noonans syndrom
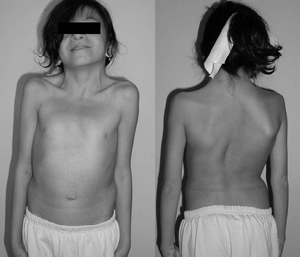
en 12-årig tjej med Syndrome
Noonans syndrom. Typisk simhud hals. Dubbel strukturell kurva med revben deformitet.,
Cardiofaciocutaneous syndrom, Turners syndrom, Costello syndrom, neurofibromatosis typ 1
baserat på symptomen
tillväxthormon
beror på svårighetsgraden av hjärtproblem
/th >
1 i 100 (1 i 2000 svår sjukdom)
Noonans syndrom (ns) är en genetisk sjukdom som kan förekomma med milt ovanliga ansiktsdrag, kort höjd, medfödd hjärtsjukdom, blödningsproblem och skelettmissbildningar., Ansiktsdrag inkluderar brett åtskilda ögon, ljusa ögon, låga öron, en kort nacke och en liten underkäke. Hjärtproblem kan innefatta lungventilstenos. Bröstbenet kan antingen sticka ut eller sänkas, medan ryggraden kan vara onormalt krökt. Intelligens i syndromet är ofta normalt. Komplikationer av NS kan innefatta leukemi.
ett antal genetiska mutationer kan resultera i Noonans syndrom. Villkoret kan vara ärvt från en persons föräldrar som ett autosomalt dominerande tillstånd eller uppstå som en ny mutation., Noonans syndrom är en typ av Rasopati, den underliggande mekanismen för vilken innefattar överaktivering inom RAS / MAPK-cellens signaleringsväg. Diagnosen kan misstänkas baserat på symtom, medicinsk bildbehandling och blodprov. Bekräftelse kan uppnås med genetisk testning.
inget botemedel mot NS är känt. Behandlingen är baserad på symptom och underliggande problem, och extra stöd i skolan kan krävas. Tillväxthormonbehandling under barndomen kan öka en drabbad persons slutliga höjd. Långsiktiga resultat beror vanligtvis på svårighetsgraden av hjärtproblem.,
uppskattningsvis 1 av 1000 personer påverkas mildt av NS, medan cirka 1 av 2000 har en svårare form av tillståndet. Män verkar påverkas oftare än kvinnor. Villkoret beskrevs först 1883 och namngavs efter amerikansk pediatrisk kardiolog Jacqueline Noonan, som beskrev ytterligare fall 1963.
tecken och symtom

onormala egenskaper hos Noonans syndrom vid 3 års ålder: notera ögonbrynslanten och vänstra ögonlocket.,

onormala egenskaper hos Noonans syndrom vid en ålder av 3 månader: notera låg-set, posteriort roteras, och onormalt bildade örat.
de vanligaste tecknen som leder till diagnosen Noonans syndrom är unika ansiktsdrag och muskuloskeletala egenskaper. Ansiktsegenskaperna är mest framträdande i spädbarn, blir mindre uppenbara med ålder hos många personer med Noonans syndrom.,
Huvud
några av de karakteristiska egenskaperna hos Noonans syndrom inkluderar ett stort huvud med överflödig hud på baksidan av nacken, låg hårlinje i nacken, hög hårlinje på framsidan av huvudet, triangulär ansiktsform, bred panna och en kort, webbad nacke.
i ögonen är hypertelorism (allmänt uppsatta ögon) en definierande egenskap, närvarande hos 95% av personer med Noonans syndrom., Detta kan åtföljas av epicanthala veck (extra hudveck vid ögonets inre hörn), ptos (hängande ögonlock), proptos (utbuktande ögon), strabismus (inåt eller utåt vridning av ögonen), nystagmus (jerking rörelse av ögonen) och brytnings visuella fel.
näsan kan vara liten, bred och uppåtvänd.
utveckling av munnen kan också påverkas i Noonans syndrom., Detta kan resultera i djupt räfflad philtrum (övre läpplinjen) (över 90%), mikrognathia (underdimensionerad underkäke), hög välvd gom, artikulationsproblem (tänder inte radas upp) vilket kan leda till tandproblem. Liknande de muskulära manifestationerna ovan, i munnen kan dålig tungkontroll observeras.,
Hud
tecken och symtom på hud i Noonans syndrom inkluderar lymfödem (lymfsvullnad i extremiteterna), keloidbildning, överdriven ärrbildning, hyperkeratos (överutveckling av yttre hudskikt), pigmenterad nevi (mörkpigmenterade hudfläckar) och bindvävssjukdom
muskuloskeletala
abnormiteter i lemmar och extremiteter kan förekomma i Noonans syndrom. Detta kan manifesteras som helt slutade fingrar, extra stoppning på fingrar och tår, ödem på baksidan av händer och toppar av fötter och cubitus valgus (bred bärande vinkel på armbågarna).,
för kortväxthet kombineras tillväxthormon ibland med IGF-1 (eller som ett alternativ, IGF-1 som fristående) för att uppnå en ökad höjd/slutlig höjd snabbare. Den slutliga vuxna höjden hos individer med Noonans syndrom är ca 161-167 cm hos män och 150-155 cm hos kvinnor, som närmar sig den nedre gränsen för normal.
spinala abnormiteter kan förekomma upp till 30% av tiden och detta kan kräva operation för att korrigera i över 60% av dessa fall., Andra muskuloskeletala manifestationer i Noonans syndrom är associerade med odifferentierade bindvävssjukdomar som kan associeras med gemensamma kontrakturer (täthet) eller gemensam hypermobilitet (löshet). Ytterligare faktorer kan förekomma i form av sveda av skulderbladet, skolios, bröst ben framträdande (pectus carinatum), bröst ben depression (pectus excavatum). Muskelavvikelser kan förekomma som hypotoni (låg muskelton), vilket kan leda till lordos (ökad ihålig i ryggen) på grund av dålig bukmuskelton.,
hjärta
Noonans syndrom är den näst vanligaste syndromiska orsaken till medfödd hjärtsjukdom. Detta inkluderar pulmonell valvulär stenos (50-60%), Förmaksseptumdefekter (10-25%), Ventrikelseptumdefekter (5-20%) och hypertrofisk kardiomyopati (12-35%).
lungor
restriktiv lungfunktion har rapporterats hos vissa personer.
Gastrointestinal
ett antal olika gastrointestinala (GI) symtom har associerats med Noonans syndrom., Dessa inkluderar sväljningssvårigheter, låg tarmmotilitet, gastroparesis (fördröjd magtömning), tarmmalrotation och frekvent eller kraftig kräkningar. Dessa matsmältningsproblem kan leda till minskad aptit, underlåtenhet att trivas från spädbarn till puberteten (75%) och ibland behovet av ett matningsrör.{ Det har associerats med Von Willebrands sjukdom, Amegakaryocytisk trombocytopeni (lågt antal blodplättar), förlängd aktiverad partiell tromboplastintid, kombinerade koagulationsfel., När det är närvarande kan dessa Noonan-syndrom som åtföljer störningar associeras med en förutsättning för blåmärken lätt eller blödning.
neurologisk
Ibland kan Chiari missbildning (typ 1) förekomma, vilket kan leda till hydrocephalus. Kramper har också rapporterats.
orsaker

NS är vanligtvis ärvt i ett autosomalt dominant mönster med variabelt uttryck.,
återfall i syskon och skenbar överföring från förälder till barn har länge föreslagit en genetisk defekt med autosomalt dominant arv och variabelt uttryck. Mutationer i Ras/mitogenaktiverade proteinkinas-signalvägar är kända för att vara ansvariga för cirka 70% av NS-Fallen.
personer med NS har upp till 50% chans att överföra den till sina avkommor., Det faktum att en berörd förälder inte alltid identifieras för barn med NS föreslår flera möjligheter:
- manifestationer kan vara så subtila att de går okända (variabel expressivitet)
- NS är heterogen, innefattande mer än ett liknande tillstånd av olika orsaker, och några av dessa kanske inte ärvs.
- en stor del av fallen kan representera nya sporadiska mutationer.,
| Typ | Online Mendelian arv i man databas | Gen | år hittades | Locus | % av fallen | beskrivning | Refs. |
|---|---|---|---|---|---|---|---|
| ns1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | PTPN11-genen kodar för proteintyrosinfosfatas SHP-2., Detta protein är en komponent i flera intracellulära signaltransduktionsvägar som är involverade i embryonal utveckling som modulerar celldelning, differentiering och migration, inklusive en medierad av epidermal tillväxtfaktorreceptorn, vilket är viktigt vid bildandet av de semilunära hjärtklaffarna. duplicering av kromosomområdet som innehåller PTPN11 kan också resultera i NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
heterozygota mutationer i NRAS, HRAS, BRAF, shoc2, map2k1, map2k2 och CBL har också associerats med en mindre andel av NS och relaterade fenotyper.
ett tillstånd som kallas ”neurofibromatos-Noonans syndrom” är associerat med neurofibromin.
diagnos
NS kan bekräftas genetiskt genom närvaron av någon av de kända mutationer som anges ovan., Trots identifiering av 14 orsakande gener kommer frånvaron av en känd mutation inte att utesluta diagnosen, eftersom mer, som-ännu-oupptäckta gener kan orsaka NS. Således är diagnosen av NS fortfarande baserad på kliniska egenskaper. Med andra ord görs det när en läkare känner att en person har tillräckligt med funktioner för att motivera etiketten. De viktigaste värdena för att göra en genetisk diagnos är att den vägleder ytterligare medicinska och utvecklingsutvärderingar, det utesluter andra möjliga förklaringar till funktionerna, och det möjliggör mer exakta återkommande riskbedömningar., Med mer genotyp-fenotyp korrelation studier som utförs, en positiv genetisk diagnos kommer att hjälpa läkaren att vara medveten om eventuella anomalier specifika för att vissa genmutation. Till exempel ses en ökning av hypertrofisk kardiomyopati hos personer med mutation av KRAS och en ökad risk för juvenil myelomonocytisk leukemi finns för en mutation av PTPN11. I framtiden kan studier leda till en riktad hantering av NS-symtom som beror på vilken genetisk mutation en person har.,
före födseln
prenatala funktioner som kan leda läkare att överväga en diagnos av Noonans syndrom inkluderar cystisk hygrom, ökad nuchal genomskinlighet, pleurautgjutning och ödem.
differentialdiagnos
medan Turners syndrom har likheter med njuranomalier och utvecklingsfördröjning, finns Turners syndrom endast hos kvinnor och uttrycker ofta annorlunda. I Turners syndrom finns det en lägre förekomst av utvecklingsförseningar, vänstersidiga hjärtfel är konstanta och förekomsten av njuravvikelser är mycket lägre.,
andra RASopathies
- Watsons syndrom – Watson syndrom har ett antal liknande egenskaper med Noonans syndrom såsom kortväxthet, pulmonell ventilstenos, varierande intellektuell utveckling och hudpigmentförändringar.
- Cardiofaciocutaneous (CFC) syndrom – CFC syndrom är mycket lik Noonans syndrom på grund av liknande hjärt-och lymfatiska egenskaper. Men i CFC syndrom intellektuella funktionshinder och gastrointestinala problem är ofta allvarligare och uttalade.,
- Costello syndrom – liknande CFC syndrom, Costello syndrom har överlappande funktioner med Noonans syndrom. Villkoren kan emellertid särskiljas av deras genetiska orsak.
- Neurofibromatosis 1 (NF1)
- Williams syndrom
hantering
behandlingen varierar beroende på komplikationer men tenderar att vara ganska standard, vilket återspeglar behandlingen av den allmänna befolkningen., Ledningsriktlinjer, dividerat med system, inklusive allmän, utvecklings, dental, tillväxt och utfodring, kardiovaskulär, audiologisk, hematologisk, njure och skelett, som tar hänsyn till åtgärder som ska vidtas vid diagnos, efter diagnos och om symptomatisk, har publicerats av ett amerikanskt konsortium.
specifikt liknar behandling av kardiovaskulära komplikationer den allmänna populationen och behandling av blödningsbenägenhet styrs av den specifika faktorbristen eller trombocytaggregationen.,
- neuropsykologisk testning rekommenderas för att hitta styrkor och utmaningar för att skräddarsy stöd som behövs för skolan och karriären.
- pedagogisk anpassning som en individualiserad utbildningsplan behövs ibland för barn i skolåldern.
- talterapi om tal – och artikulationsproblem förekommer
- fysisk terapi och arbetsterapi för brutto-och finmotoriska förseningar
- hypotoni och motoriska svårigheter påverkar ofta handstil. Boende för att minska handskriftsbehov kommer att förbättra prestanda och spara långsiktig handfunktion.,
- periodisk uppföljning och livslång övervakning av abnormiteter som finns i något system, särskilt kardiovaskulärsystemet, rekommenderas.
anestesi risk
även om ett fåtal personer med Noonans syndrom har rapporterats utveckla malign hypertermi, är genmutationen av sjukdomar som är kända för att vara associerade med malign hypertermi annorlunda än för Noonans syndrom.,
prognos
livslängden för personer med Noonans syndrom kan likna den allmänna befolkningen, men Noonans syndrom kan associeras med flera hälsoförhållanden som kan bidra till dödligheten. Den största bidragsgivaren till dödligheten hos individer med Noonans syndrom är komplikationer av hjärt-kärlsjukdom. Prognosen är därför till stor del beroende av närvaron eller frånvaron av hjärtsjukdom, liksom typen och svårighetsgraden av sjukdomen (om sjukdomen är närvarande)., I synnerhet Noonans syndrom med hypertrofisk kardiomyopati associerad med ökad dödlighet.
historia
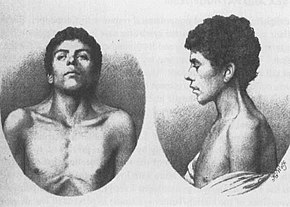
det äldsta kända fallet av NS, beskrivet 1883 av Kobylinski
Jacqueline Noonan övade som pediatrisk kardiolog vid University of Iowa när hon märkte att barn med en sällsynt typ av hjärtfel, var det möjligt att valvulär pulmonell stenos, ofta hade en karakteristisk fysiskt utseende, med kort resning, simhud hals, breda åtskilda ögon, och låg-set öron., Både pojkar och flickor påverkades. Dessa egenskaper sågs ibland i familjer men var inte förknippade med grova kromosomala abnormiteter. Hon studerade 833 personer med Noonans syndrom vid medfödd hjärtsjukdom klinik, letar efter andra medfödda abnormiteter, och 1963 presenterade ett papper: ”associerade icke-hjärtmissbildningar hos barn med medfödd hjärtsjukdom”. Detta beskrev 9 barn som förutom medfödd hjärtsjukdom hade karakteristiska ansiktsdrag, bröstdeformiteter och kortväxthet.
Dr. John Opitz, en före detta student av Dr., Noonans, började först kalla tillståndet ”Noonans syndrom” när han såg barn som såg ut som de som Dr Noonan hade beskrivit. Dr. Noonan producerade ett papper med titeln ”Hypertelorism with Turner Phenotype” 1968 där hon studerade 19 patienter som visade symptom som indikerar Noonans syndrom. 1971 vid symposiet av kardiovaskulära defekter blev namnet ”Noonans syndrom” officiellt erkänt.
















© 2021 Tombouctou
Tema av Anders Noren — Upp ↑
Lämna ett svar