introduktion
intellektuell funktionsnedsättning (ID) påverkar nästan 1-2% av befolkningen och är den vanligaste neurodevelopmental oordning (NDD). Ett betydande antal ID-patienter visar sig ha en genetisk orsak (granskad i Bessa et al., 2012)., Genomet-omfattande analystekniker som för närvarande används för undersökning av etiologi leder ofta till identifiering av mycket sällsynta nästan privata varianter, insamling av patienter med förändringar i samma gen är en avgörande aspekt av definitionen av en ny klinisk enhet.
tidigare i år har patienter som hyser mutationer i ebf3-genen beskrivits och presenterar ett neurodevelopmental syndrom inklusive ID, ataxi, hypotoni, mild ansiktsdysmorfism och genitourinary abnormalities (OMIM 617330) (Chao et al., 2017, Skadar et al., Och med 2017. Sleven et al., 2017)., Ebf3-genen (tidig B-Cellfaktor 3) kodar för en medlem av den högt konserverade tidiga B-cellfaktortranskriptionsfaktorfamiljen, uttryckt vid höga nivåer i det utvecklande nervsystemet (data hämtad från Gtex-portalen). EBF3 är en transkriptionell mål i ARX, och visat sig vara reglerad av NeuroD och ARX (Friocourt och Parnavelas, 2011). ARX kodar en transkriptionsfaktor som är kritisk för embryonal utveckling som i många år har förknippats med ett brett spektrum av neurodevelopmental disorders., Den intellektuella försämringen, centrala nervsystemet och genitourinära anomalier som observerats hos patienter med både mutationer i EBF3 och ARX kan återspegla båda proteins bidrag till samma molekylära och cellulära processer (Chao et al., 2017). EBF3-funktionen har också studerats i djurmodeller. Ablation av dess ortologer i maskar och flugor leder till försämring av neuronal utveckling (Prasad et al., 1998; Hattori et al., 2013)., Hos möss, knackar ut Ebf3 leder till neonatal dödlighet och neuronala migrationsdefekter, med fel på luktneuroner projekt till dorsal olfaktorisk glödlampa (Wang et al., 2004). De exakta patogena mekanismerna för ebf3-mutationer är ännu inte helt klarlagda, men den typ av varianter som hittills beskrivits tyder på att haploinbrist, funktionsförstärkning och dominerande negativ är möjliga patogena mekanismer för de beskrivna varianterna (Chao et al., Och med 2017. Sleven et al., 2017).,
i detta arbete bidrar vi med en patient med den minsta raderingen (600 Kb) som hittills rapporterats påverka totaliteten av ebf3-genen och med en klinisk presentation som överlappar den hos patienter med Ebf3 enstaka nukleotidvarianter (SNVs). Dessutom gör vi en klinisk jämförelse av patienterna med tidigare publicerade stora terminal 10q-deletioner och rapporterar att det trots skillnaderna i storlek finns en signifikant fenotypisk överlappning mellan patienter med dessa förändringar., Dessa fynd lägger till den nuvarande kunskapen om EBF3-relaterade störningar och stöder EBF3 haploinbrist som nyckel i neurodevelopmental syndrom associerat med 10qter-deletioner.
material och metoder
patienten fastställdes inom en stor studie av neurodevelopmental disorders i Portugal, Där inskrivning av patienter och familjer gjordes av den hänskjutande läkaren, klinisk information samlades in i en anonym databas enligt den portugisiska dataskyddsmyndigheten (CNPD) och skriftligt informerat samtycke erhölls för alla deltagare., Informerat samtycke till den nuvarande patienten tillhandahölls av moderen för den genetiska studien och publiceringen av resultat (inklusive bilder). Studien godkändes av den etiska kommittén för Centrum för Medicinsk Genetik Dr Jacinto Magalhães, National Health Institute Dr. Jorge Ricardo.
Genomiskt DNA extraherades från perifert blod med hjälp av antingen Citogene® DNA-isoleringskit (Citomed, Portugal). aCGH utfördes med hjälp av Agilent 180 K array (AMADID:023363) mot en diploida DNA-referens (Kreatech är MegaPoll Referens-DNA, Kreatech Diagnostik, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., Analysen utfördes i en 7500-snabb PCR-maskin i realtid (Applied Biosystems, Foster City, CA, USA) med hjälp av Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) enligt tillverkarens rekommendationer och enligt de allmänna riktlinjerna för qPCR. Specificiteten för varje reaktion verifierades genom generering av en smältkurva för var och en av de förstärkta fragmenten. Primereffektiviteten beräknades genom generering av en standardkurva som passar den accepterade normala effektivitetsprocenten (primrar som används i tilläggsdata)., Ct-värden som erhålls för varje prov analyserades i DataAssist™ programvara (Applied Biosystems, Foster City, CA, USA).
resultat
här beskriver vi en patient med en de novo-radering som påverkar EBF3. Patienten är en 11 – årig tjej med svår ID (global development quotient = 27 vid 7 års ålder), född från icke-konsanguineösa föräldrar och utan familjehistoria av neurodevelopmental disorders. Hon föddes efter en biamniotisk bichorionisk tvillinggraviditet (hennes syster är frisk), genom vaginal leverans, vid 35 veckors graviditet. Födelseparametrar var: vikt, 1,830 g (P3); Längd, 42.,5 cm (P10); och OFC, 30,6 cm (P10), med en Apgar poäng på 8/9 (1: A och 5: e min, respektive). Neonatalperioden var komplicerad med sepsis och diagnosen ärftlig spherocytosis (ärvd från sin mamma). Global utvecklingsfördröjning noterades under de första månaderna, med huvudkontroll uppnådd vid 12 månader, sittande vid 18 månader, oberoende promenad vid 30 månader och inga ord som talas vid 3 års ålder., Hon hade pyelonefrit vid 19 månader (renal ultraljud visade inga abnormiteter), gastroesofageal reflux och återkommande otitis media, med ledande hörselnedsättning som krävde kirurgisk ingrepp och hörapparat. Epilepsi misstänktes vid 5 månader (episoder av suspenderad aktivitet) men EEG var normalt.
hon observerades först vid 3 års ålder 5 månader (figurerna 1A,B), vid vilken tidpunkt hon visade muskelhypotoni, hypotonisk ansikte, strabismus och minskad känslighet för smärta., Hon presenterade också milda dysmorfa egenskaper (Figur 1A): triangulärt ansikte, små låga öron med framträdande Anti-helix, välvda ögonbryn, anteverted nares, bulbous nasal spets, liten mun med nedåtvända hörn, spetsig haka, kort hals och framträdande fingerfosterkuddar, samt en mild kort Storlek (89 cm, motsvarande cirka 2SD).

Figur 1. (A) patientens ansiktsutseende vid 3 år och 5 månader som visar de små och låga öronen med framträdande Anti-helix och (B) fosterkuddar i fingrarna., C) patientens ansiktsutseende vid 11 års ålder. (D) Markeras i grått 600 Kb radering på 10q26.3-regionen, en zoom i EBF3-genen i DGV-databasen visar att det finns 3 strykningar i 3 kontroller som påverkar de första 6 exoner av EBF3 (NM_001005463); CNVs inom denna region finns i kontroll populationer har strykningar nvs825626 (finns i 1/31 individer), nvs552315 (finns i 1/17421 individer) och nsv552316 (finns i 1/31 individer). (E) schematisk representation av EBF3 avskrifter.,
HJÄRNMRI utfördes vid 6 år, men inga abnormiteter noterades. Vid 10 års ålder omvärderades hon; hon hade fortfarande återkommande otitis media, men annars var hon i god global hälsa. Språket var mycket dåligt (två ord meningar som talas efter 8 år). Hon hade beteendeproblem, med stereotypa rörelser (roterande rörelser, tugga på kläder, Huvud retropulsion), scoring för svår autism spectrum disorder (ADI-r och ADOS) vid 7 år; hon visade agitation och aggressivt beteende (auto och hetero) och medicinerades med antipsykotiska läkemedel., En ortopedisk kirurgi utfördes för pes planus. Ansiktsegenskaperna liknade de som tidigare beskrivits, med fördelade övre centrala snedställningar (figur 1C); hon hade glasögon för strabismus och hypermetropi.
Analys av genomiskt DNA av aCGH visade en de novo 600 kb radering på 10p26.3 (Figur 1D) som påverkar tre gener—MGMT (kodning av enzymet O-6-methylguanine-DNA metyltransferas, som är involverade i DNA-reparation), EBF3 och GLRX (kodning glutaredoxin, en liten thioltransferase som tar bort protein GSH adducts), som EBF3 var den mest sannolika sjukdom-associerade gener.,
diskussion
den presenterade patienten analyserades först av aCGH för några år sedan. Vid tidpunkten för aCGH-analys, förekomsten av de tre varianterna som finns i databasen av genomiska varianter (DGV)1 som påverkar de första fem exonerna av ebf3-genen (figur 1e; Park et al., 2010, Cooper m.fl., 2011), liksom frånvaron av andra kända sjukdomar som orsakar mutationer i denna gen, leder oss att klassificera det som en variant av okänd betydelse (VOUS). Men de senaste publikationerna av EBF3 mutationer (Chao et al., 2017, Skadar et al., Och med 2017. Sleven et al.,, 2017) och de kliniska likheterna med de rapporterade fallen leder oss till att ompröva varianten och få oss att tro att ebf3-radering faktiskt kan redovisa sjukdomen hos patienten. En av de aspekter som väckte tvivel om patogeniciteten hos denna variant i första hand var förekomsten av befolkningskontroller med deletioner av de första sex exonerna av denna gen, i heterozygositet (data hämtade från DGV-databasen från och med februari 2017) (figur 1D). Även om en avskrift av EBF3 börjar i Exon13b anges i Ensembl-databasen (ENST00000440978.,1) (figur 1E), som kan förklara hur radering av de första exonerna så småningom skulle kunna resultera i en normal fenotyp, utesluter detta transkript EBF3: s DNA-bindande domän, och dess uttrycksmönster och funktionell relevans har inte karakteriserats. Vid omprövning, dock CNVs beskrivs av Cooper och kollegor (nsv552315 och nsv552316) (Chao et al., 2017, Skadar et al., Och med 2017. Sleven et al.,, 2017) ansågs ligga på tröskeln för detektion av SNP microarray och kan inte ligga till grund för uteslutning av en kandidatgen, särskilt mot bakgrund av de starka genetiska och funktionella bevisen för EBF3-mutations relevans för sjukdom (Evan Eichler, Greg Cooper och Bradley Coe, personlig kommunikation).,
vår patient visar många kliniska likheter med tidigare beskrivna patienter med mutationer i EBF3 (21 fall sammanfattade i Tabell 1), såsom global utvecklingsfördröjning, fördröjd uttrycksfull tal, hypotoni, ökad smärttröskel, beteendeproblem och karakteristiska ansiktsdrag (lång/triangulär ansikte, stor panna, hypoton ansikte)., Men även om vår patient hade betydande fördröjning i motorisk utveckling, detekterades ingen signifikant ataxi (med vissa begränsningar i klinisk undersökning, eftersom barnet inte var kooperativt) och inga cerebellära anomalier var närvarande i HJÄRNMRI.

tabell 1. Klinisk jämförelse av det aktuella fallet med de rapporterade fallen med punktmutationer / Indel i ebf3-genen.
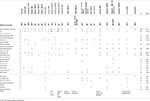
tabell 2., Klinisk jämförelse av det aktuella fallet med de rapporterade fallen med deletioner som påverkar 10q26 cytoband.
EBF3-varianterna som beskrivs i litteraturen i början av 2017 inkluderar punktmutationer som förutspås vara skadliga och små Infogningar och borttagningar som leder till att viktiga aminoacider raderas eller till en frameshift, vilket förutsägbart orsakar tidig trunkering av det resulterande proteinet eller nonsensmedierade sönderfallet., Mutationerna koncentrerades på delar av genen som kodade den DNA-bindande domänen av EBF3, och förutspåddes genom olika metoder för att leda till en förlust av funktion av denna transkriptionsfaktor, vilket tyder på minskad funktion och haploinsufficiens som mekanismen bakom neurodevelopmental störning hos dessa patienter. Knock-out möss för Ebf3 beskrivs för att presentera neonatal dödlighet och neuronala migrationsdefekter, med misslyckande av olfaktoriska neuroner för att projicera till dorsal olfaktorisk glödlampa (Wang et al.,, 2004), men ingen beskrivning är gjord av en fenotyp i de heterozygota djuren, som faktiskt presenteras som kontroller i många av experimenten, vilket inte stöder haploinmissbruksmodellen. Vi gjorde ansträngningar för att få och studera dessa djurs neurodevelopmental fenotyp, men lyckades inte, eftersom Ebf3 (O/E2) knock-out-linjen kan ha avbrutits (Joseph W. Lewcock, personlig kommunikation). Det nuvarande fallet tillsammans med patienterna som sammanfattas i Tabell 2 stöder emellertid hypotesen om EBF3 haploinbrist som sjukdomsframkallande.,
Sammanfattningsvis förstärker den nuvarande beskrivningen ebf3 förlust av funktion / haploinbrist som orsak till neurodevelopmental sjukdom och förstärker föreningen av denna gen med ett karakteristiskt kliniskt syndrom inom detta spektrum.
Författarbidrag
FL utförde molekylära studier och analyserade molekylära data. MG och GS samlade in kliniska data. FL, JP och PM granskade alla ebf3 mutation fall i litteraturen. FL, GS, JP och PM utarbetade tidningen. PM erhöll finansiering för denna studie. Studien utfördes under PM-riktningen.,
Finansieringen
FCT—Fundação para en Ciência e Tecnologia inom projekt och stipendier (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Denna artikel har utvecklats inom ramen för projektet NORTE-01-0145-FEDER-000013, med stöd av norra Portugals regionala operativa program (NORTE 2020), enligt partnerskapsavtalet Portugal 2020, genom Europeiska regionala utvecklingsfonden (FEDER).
intressekonflikt uttalande
JP var anställd av företaget CGC Genetics.,
de andra författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
bekräftelser
vi vill tacka patienten och hennes familj för deltagande i de genetiska studierna och för att tillåta denna publikation. Vi är tacksamma mot Dr Ana Maria Fortuna och Dr Margarida Reis Lima för att göra detta projekt möjligt och genom att underlätta vårt samarbete med CGM.
fotnoter
1., ^Databas med genomiska varianter finns på: http://dgv.tcag.ca/dgv/app/home .
Friocourt, G., och Parnavelas, J. (2011). Identifiering av Arx-mål avslöjar nya kandidater för att kontrollera kortikal interneuron migration och differentiering. Front. Cell. Neurosci. 5:28. doi: 10.3389/fncel.2011.00028
PubMed Abstrakt | CrossRef Full Text | Google Scholar
















Lämna ett svar