OSS Pharm. 2010;35(5):36-41.
En sammenheng mellom diabetes mellitus (DM) og demens er unektelig, med en rekke studier konkluderer med at DM øker risiko for kognitiv svikt og demens, inklusive Alzheimers sykdom (AD).1-5 Ikke bare DM øke risikoen for demens, er det faktisk øker frekvensen av demens utvikling i to – tre ganger.,3 mekanismen for dette verdifallet ikke er fullt ut forstått, men det er en hypotese om at hyperglykemi, insulinresistens, oksidativt stress, avansert glycation end-produkter, og inflammatoriske cytokiner kollektivt føre til kognitiv dysfunksjon.5 I faktum, diabetes ble beskrevet som en «spesiell form for akselerert aldring» i 1976 på grunn av sine mange tilhørende komplikasjoner.,5 Den tilsynelatende overlapp mellom DM og demens har ført til forslag om at ANNONSEN er ikke utelukkende en nevrologisk lidelse, men heller en nevroendokrine lidelse, med Steen et al coining begrepet type 3 diabetes for å beskrive denne hybrid sykdom.,6
Oversikt over insulinresistens/Nedsatt Glukose Toleranse
Pathophysiology: Både nedsatt glukosetoleranse—definert som en plasma-glukose på 140 mg/dL til 199 mg/dL etter en oral glukose toleranse test—og impaired fasting glucose—definert som en fastende plasma glukose 100 mg/dL til 125 mg/dL—er diagnostiske av prediabetes (forstadium til type 2 DM ), som er en hemoglobin A1C på 5,7% til 6,4 prosent.7 pathophysiology av prediabetes er den samme som for T2DM, som er diagnostisert etter et fastende blodsukker på 126 mg/dL eller mer eller et A1C av 6.,5% eller mer (testen gjentas for bekreftelse).7 I begge forhold, insulinresistens utvikler seg i perifere vev, der insulin er nødvendig for at glukose opptak (nemlig muskel, lever og fett), og bukspyttkjertelen er tvunget til å levere stadig mer insulin for å overvinne denne motstanden.8 I denne fasen, pasienter er ofte hyperinsulinemic, men over tid, bukspyttkjertel ikke kan oppfylle kravene i vevet, og de insulin-produserende beta-cellene i bukspyttkjertelen sakte mislykkes å produsere stadig mindre insulin.,8 Uten tilstrekkelig insulin, blodsukker konsentrasjoner stige, noe som fører til prediabetes og, i opp til 70% av pasienter med prediabetes, T2DM.9 komplikasjoner av hyperglykemi er mange og omfatter en økt risiko for macrovascular komplikasjoner (inkludert hjerteinfarkt, cerebrovaskulær, og perifer vaskulær sykdom) og microvascular komplikasjoner (inkludert nefropati, neuropathy, og retinopati).7
Behandling: Mange behandlinger finnes for å behandle DM., Den innledende behandling for både prediabetes og T2DM er en livsstil endring som involverer kosthold, mosjon og vekttap.7 The American Diabetes Association (ADA) anbefaler også hensynet til metformin hos pasienter med prediabetes som har høy risiko for å utvikle diabetes.7 Metformin er anbefalt som første farmakologiske behandling for T2DM.7 Ofte, flere farmakologiske behandlingen er nødvendig for å oppnå ADA glykemisk mål, inkludert en A1C <7%.,7 Det er mange klasser av medisiner for T2DM (TABELL 1), og valg av representant til å normalisere blodsukkeret, avhenger av en rekke pasient – og medikament-spesifikke faktorer. Det er viktig å merke seg at mange pasienter med T2DM vil eventuelt kreve eksogene insulin for å oppnå og opprettholde euglycemia som beta-celle funksjon gradvis avtar.
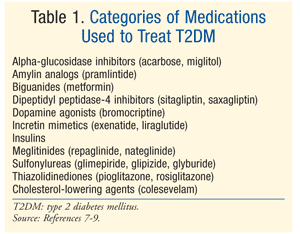
Oversikt over ANNONSEN
Pathophysiology: AD er den vanligste formen for demens hos eldre voksne, sto for 60% til 80% av alle tilfeller.,10 To histopathologic kjennetegnene på ANNONSEN er neuritic plaketter og neurofibrillary floker.11 Plakk består av uløselige beta-amyloid protein. Floker, som er intracellulære, er sammensatt av phosphorylated tau-protein. Tau-protein er viktig for microtubule forsamlingen, og når dette proteinet er unormalt phosphorylated, neuronal funksjon er avbrutt. Neurofibrillary floker som oftest påvirke kolinerge nevroner.12 I tillegg, forstyrrelser av de viktigste excitatory nevrotransmitter, glutamat, bidrar til patologi av ANNONSEN., I ANNONSEN er det en overactivation av glutamat, signaliserer en prosess som er kjent som excitotoxicity, noe som fører til dannelsen av plakk, hyperphosphorylation av tau-protein, og celledød.12 I sammendraget, excitotoxicity og dannelse av neuritic plaketter og neurofibrillary floker forstyrre nevrotransmitter trasé, noe som resulterer i læring og hukommelse verdifall knyttet til ANNONSEN.
Behandling: De tilgjengelige FDA-godkjent farmakologiske behandlinger for AD er oppsummert i TABELL 2.,11,12 Mens det fortsatt er mye å lære med hensyn til behandlinger som behandle eller endre patologi ANNONSE, for tiden tilgjengelig terapi involverer bruk av acetylkolinesterase-hemmere, som det er fire, og N-metyl-d-aspartat-reseptor antagonist memantine.

Insulin og Hjernen
Selv om det er nødvendig for at glukose transport i perifere vev, insulin ser ikke ut til å være nødvendig for transport av glukose inn i hjernen eller for cerebral glukose metabolisme., Mens glukose transport til hjernen er ikke avhengig av insulin, insulin i seg selv er gjennomført over blod–hjerne-barrieren (BBB) med insulin reseptor–mediert transport prosesser.13 Dette transport mekanisme er saturable; på lengre perioder med overflødig insulin konsentrasjoner (hyperinsulinemia) i periferien, slik som de man ser i prediabetes og T2DM, dette reseptor-mediert transport mekanisme downregulates, og dermed redusere transport av insulin i hjernen og spinalvæske.,
I tillegg til insulin, insulin-like growth factor type 1 (IGF-1) er til stede i hjernen, og er nødvendig for normal vekst og funksjon av sentralnervesystemet (CNS).14 Insulin og IGF-1-reseptorer er plassert over hele hjernen på nevroner og astrocyttene (stjerne-formet celler i CNS som bidrar til å støtte nevroner i hjernen). To områder av hjernen som er viktig for læring og hukommelse, hippocampus og hypothalamus, inneholder høye konsentrasjoner av insulin reseptorer.5 I rotter, intracerebroventricular insulin administrasjon har vist seg å øke minne.,15 I tillegg, intranasal insulin administrasjon i mennesker har blitt funnet å øke minne ytelse.16 Som tidligere nevnt, er insulin er ikke nødvendig for cerebral glukose-metabolisme; imidlertid kan det være spesielle områder i hjernen som insulin utløser metabolske prosesser som involverer glukose. I rotte studier, insulin har vist seg å påvirke glukose utnyttelse i hypothalamus og locus coeruleus, to områder som er viktige for læring og hukommelse.,17 Læring har også vist seg å legge til rette for økt uttrykk av CNS insulin reseptorer, som fører til postulering som tilstedeværelse og aktivitet av insulin i hjernen bidra til, og spille en viktig rolle i læring og hukommelse. Insulin kan også bidra til at modulering av CNS nevrotransmittere, særlig acetylkolin og noradrenalin, som begge er kritisk for normal kognisjon og hjernen helse.,18
insulinresistens og Demens
Oversikt: Det synes å være mange feil i insulin signalering i hjernen av ANNONSEN pasienter, noe som fører til redusert glukose utnyttelse og energimetabolisme.6,19 Som T2DM er forbundet med perifer insulinresistens, AD er forbundet med hjernen insulinresistens.20 Tidlig i ANNONSEN, det synes å være utilstrekkelig insulin opptak og signaler i hjernen, et tegn på insulinresistens.5,19,21,22 Økt insulin reseptorer har blitt sett i hjernen til pasienter med AD, sannsynligvis som en kompensasjon for insulinresistens.,21
Samlet sett gir dette verdifall i insulin signaliserer har mange nedstrøms effekter i ANNONSEN. En teori er at lave konsentrasjoner av insulin i CNS føre til en reduksjon i acetylkolin nivåer og cerebral blodstrøm.13 i Tillegg, endringer i insulin konsentrasjoner kan fremme beta-amyloid og tau-protein-formasjonen.2 I hjernen, overskotsenergi (IDE) er involvert i nedbrytning og rydding av beta-amyloid protein.21 Høye nivåer av insulin hemmer IDE og kan føre til en nedgang i beta-amyloid klaring, deretter økende hjernen deponering av beta-amyloid.,5,19,21
Nylig ceramides, en familie av lipider, har befalt oppmerksomhet for sine potensielle rolle i insulinresistens og demens.23,24 Ceramides er generert i nærvær av betennelse, noe som er vanlig i fedme, T2DM, og AD.24 Fordi ceramide lett krysser BBB, eksponering fører til nedsatt energi metabolisme og endringer i insulin genuttrykk, og bidrar til insulinresistens.23,24 Dette er for tiden et aktivt område av interesse, med forskere som viser at hemme ceramide syntese forhindret fedme-mediert insulinresistens.,24
Intranasal Insulin: Som tidligere nevnt, intranasal insulin administrasjon forbedrer minne ytelse i mennesker.16 Flere studier av intranasal insulin har blitt gjennomført. Ved administrasjon av 20 enheter av intranasal insulin to ganger daglig for å menneskelig fag, Reger et al fant at, sammenlignet med placebo-gruppen behandlet med insulin beholdt mer verbal informasjon og viste økt oppmerksomhet og funksjonell status.,25 av Disse forskerne har også bemerket at fastende plasma glukose og insulin ikke endre med intranasal insulin bruk, noe som indikerer at intranasal administrering gir direkte insulin tilgang til CNS uten ytre effekter.25,26
PPAR-Gamma-Agonister: Grundig studert for behandling av perifer insulinresistens, det thiazolidinediones rosiglitazone og pioglitazon blir etterforsket for sin rolle i behandling av ANNONSEN. Thiazolidinediones er peroxisome proliferator-aktivert reseptor (PPAR)–gamma-agonister., PPAR-gamma er involvert i glukose og lipid metabolisme og modulerer betennelse, en kjent bidragsyter til AD.22,27 Thiazolidinediones er insulin sensitizers, arbeider for å redusere insulin motstand; de vises også til å redusere betennelse.21,26 I flere placebo-kontrollerte studier, pasienter behandlet med rosiglitazone viste bedre hukommelse og kognitiv funksjon.21,22,28 Selv om bare små studier av pioglitazone har blitt gjennomført, denne medisinen har også vært assosiert med forbedringer i hukommelse og kognisjon.,22 Selv om pioglitazone og rosiglitazone har dårlig BBB permeabilitet, og hver av disse midlene har blitt dokumentert i hjernen etter oral administrasjon.22 Selv om disse medikamentene kan spille en rolle i forebygging eller behandling av AD, de er for øyeblikket ikke en FDA-godkjent for pasienter med AD, og deres mulige fordeler og risikoer må være grundig vurdert på individuelt grunnlag.
Aerob Trening: Kanskje det mest effektive, og de mest oversett metode for å redusere insulin resistens er fysisk aktivitet., Fedme har vært forbundet med en økt risiko for insulinresistens, diabetes, demens, og AD.29,30 Trening forbedrer insulinfølsomhet og senker perifer insulin nivåer.26,31 Fysisk aktivitet, uavhengig av vekt, er forbundet med en lavere risiko for AD, noe som gjør det til det bare påvist neuroprotective terapi.29-31 En mulig mekanisme av dette neuroprotection er at øvelsen fremmer klarering av beta-amyloid proteiner og øker hjernen-avledet neurotrophic factor, en vekst faktor avgjørende for erkjennelse og neuronal overlevelse som er redusert i ANNONSEN.,32
Apotek Rolle
Farmasøyter spille en viktig rolle i å utdanne pasienter om nye behandlingsformer; de må også holde seg oppdatert på nye medisinske oppdagelser eller forhold som ofte presenteres i nyhetene. Type 3 diabetes er en slik tilstand. Mens det fortsatt er mye å lære om denne potensielle nye koblingen mellom diabetes og nedsatt kognisjon, det offentlige vil være å spørre sine helsepersonell’ mening. I tillegg har mange pasienter med diabetes vil være interessert i å lære mer, og vil slå til sine farmasøyter for informasjon., Derfor, farmasøyter bør diskutere den gjeldende forståelsen av denne tilstanden med sine pasienter og også rådføre seg med dem om strategier som kan redusere risikoen, gitt deres individuelle comorbidities.
Konklusjon
Intensive studier er underveis i et forsøk på å bedre preger type 3 diabetes og for å utvikle forebyggende og terapeutiske strategier. Til dags dato, det er ingen spesifikk behandling med dokumentert effekt i forebygging av kognitiv svikt eller AD hos pasienter med DM., Derfor, behandling av kognitiv svikt hos pasienter med DM er den samme som for pasienter uten DM, med spesiell oppmerksomhet til glykemisk kontroll og kardiovaskulære risikofaktorer. Dette er samme tilnærming som allerede er avgjørende for å hindre DM-relaterte komplikasjoner. Mens noen pasientgrupper synes å være økt risiko for kognitiv svikt, de fleste pasienter med DM ikke har godt definerte kliniske foreninger., Selv om ingen spesifikk behandling alternativer er tilgjengelige, gjeldende logisk tilnærming er å oppnå glykemisk kontroll og saklig administrere kardiovaskulære risikofaktorer, slik som hyperlipidemi og hypertensjon.
1. Stewart R, Liolitsa D. Type 2 diabetes mellitus, kognitiv svikt og demens. Diabet Med.
2. Xu WL, von Strauss E, Qiu CX, et al. Ukontrollert diabetes øker risikoen for Alzheimers sykdom: en populasjonsbasert kohort studie. Diabetologia. 2009;52:1031-1039.
3. Arvanitakis Z, Wilson RS, Bennett DA. Diabetes mellitus, demens og kognitiv funksjon hos eldre personer., J Nutr Helse Aldring. 2006;10:287-291.
4. Jacobson AM, Musen G, Ryan CM, et al. Langsiktig effekt av diabetes og dens behandling på kognitiv funksjon. N Engl J Med. 2007;356:1842-1852.
5. Whitmer RA. Type 2 diabetes og risiko for kognitiv svikt og demens. Curr Neurol Neurosci Rep. 2007;7:373-380.
6. Steen E, Terry BM, Rivera EJ, et al. Nedsatt insulin og insulin-like growth factor uttrykk og signaler mekanismer i Alzheimers sykdom—er denne type 3 diabetes? J Alzheimers Dis. 2005;7:63-80.
7. American Diabetes Association. Standarder for medisinsk behandling i diabetes—2010., Diabetes Care.
– 8. Triplitt Cl, Reasner CA, Isley WL. Diabetes mellitus. I: DiPiro JT, Talbert RL, Yee GC, et al, red. Farmakoterapi Prinsipper & Praksis. 7. utgave. New York, New York: McGraw Hill Medisinske; 2008:1205-1241.
9. Nathan DM, Buse JB, Davidson MD, et al. Medisinsk behandling av hyperglykemi i type 2 diabetes: en konsensus algoritme for initiering og justering av behandling: en konsensus uttalelse av American Diabetes Association og European Association for the Study of Diabetes. Diabetes Care.
10. Hebert LE, Scherr PA, Bienias JL, et al., Alzheimers sykdom sykdom i den AMERIKANSKE befolkning: prevalensestimater bruker-og boligtellingen 2000. Arch Neurol. 2003;60:1119-1122.
11. Cummings J. Alzheimers sykdom. N Engl J Med. 2004;351:56-67.
12. Slattum PW, Swerdlow RH, Hill ER. Alzheimers sykdom. I: DiPiro JT, Talbert RL, Yee GC, et al, red. Farmakoterapi Prinsipper & Praksis. 7. utgave. New York, New York: McGraw Hill Medisinske; 2008:1051-1065.
13. Håndverket S, Watson GS. Insulin og nevrodegenerativ sykdom: felles og spesifikke mekanismer. Lancet Neurol. 2004;3:169-178.
14. de la Monte SM, Tryllestaver JR., Gjennomgang av insulin og insulin-like growth factor uttrykk, signalering, og svikt i det sentrale nervesystemet: relevans i forhold til Alzheimers sykdom. J Alzheimers Dis. 2005;7:45-61.
15. Park CR, Seeley RJ, Craft S, Skogen SC. Intracerebroventricular insulin øker minne i en passiv-unngå oppgave. Physiol Behav. 2000;68:509-514.
16. Fehm HL, Perras B, Smolnik R, et al. Manipulere neuropeptidergic trasé i mennesker: en ny tilnærming til neuropharmacology? Eur J Epidemiologically. 2000;405:43-54.
17. Zhao WQ, Alkon DL. Rollen som insulin og insulin reseptor i læring og hukommelse., Mol Cell Endocrinol. 2001;177:125-134.
18. Kopf S, Baratti C. Virkninger av posttraining administrasjon av insulin på oppbevaring av en tilvenning svar på mus: deltakelse av en sentral kolinerg mekanisme. Neurobiol Lære Mem.
19. Revill P, Moralske MA, Prous JR. Nedsatt insulin signalering og patogenesen av Alzheimers sykdom. Legemidler Som I Dag (Barc). 2006;42:785-790.
20. Frost N, Tong M, Longato L, et al. Begrenset Alzheimers sykdom-type neurodegeneration i eksperimentell fedme og type 2 diabetes mellitus. J Alzheimers Dis. 2008;15:29-44.
21. Haan MN., Terapi innsikt: type 2 diabetes mellitus og risikoen for sent-Alzheimer. Nat Clin Pract Neurol. 2006;2:159-166.
22. Landreth G, Jiang Q, Madrekar S, et al. PPARgamma agonister som therapeutics for behandling av Alzheimers sykdom. Neurotherapeutics. 2008;5:481-489.
23. Lyn-Cook LE, Lawton M, Tong M, et al. Nedsatt ceramide kan megle hjernen insulinresistens og neurodegeneration i type 2 diabetes og alkoholfrie steatohepatitis. J Alzheimers Dis.
24. Tang M, de la Monte SM. Mekanismer av ceramide-mediert neurodegeneration. J Alzheimers Dis.
25., Reger MA, Watson GS, Grønn PS. Intranasal insulin forbedrer erkjennelse og modulerer beta-amyloid i begynnelsen av ANNONSEN. Nevrologi. 2008;70:440-448.
26. Håndverket S. insulinresistens syndrom og Alzheimers sykdom: alder – og fedme-relaterte virkninger på hukommelse, amyloid og betennelse. Neurobiol Aldring. 2005;26(suppl 1):65-69.
27. Jiang Q, Heneka M, Landreth GE. Rollen som peroxisome proliferator-aktivert reseptor-gamma (PPARgamma) i Alzheimers sykdom: terapeutiske implikasjoner. CNS Narkotika. 2008;22:1-14.
28. Watson GS, Cholerton BA, Reger MA, et al., Bevart erkjennelse hos pasienter med tidlig Alzheimers sykdom og amnestic mild kognitiv svikt under behandling med rosiglitazone: en forstudie. Am J Geriatr Psychiatry. 2005;13:950-958.
29. Luchsinger JA, Mayeux R. Adiposity og Alzheimers sykdom. Curr Alzheimers Sykdom Res. 2007;4:127-134.
30. Whitmer RA. Epidemiologi adiposity og demens. Curr Alzheimers Sykdom Res. 2007;4:117-122.
31. Watson GS, Craft S. rollen av insulinresistens i patogenesen av Alzheimers sykdom: implikasjoner for behandling. CNS Narkotika. 2003;17:27-45.
32. Cole GM, Frautschy SA., Rollen som insulin og neurotrophic factor signalering i hjernens aldring og Alzheimers sykdom. Exp Gerontol. 2007;42:10-21.
















Legg igjen en kommentar