Mannlige Turner syndrom, Noonan-Ehmke syndrom, Turner-lignende syndrom, Ullrich-Noonan syndrom
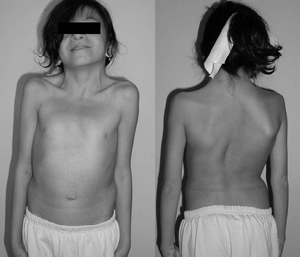
En 12-år gammel jente med Noonan syndrom. Typisk svømmehud halsen. Dobbeltrom strukturelle kurve med rib misdannelse.,irmed med genetisk testing
Cardiofaciocutaneous syndrom, Turner syndrom, Costello syndrom, nevrofibromatose type 1 –
– >
Basert på symptomer
veksthormon
kommer An på alvorlighetsgraden av hjerteproblemer
1 i 100 (1 i 2,000 alvorlig sykdom)
Noonan syndrom (NS) er en genetisk lidelse som kan presentere med mildt uvanlig ansiktstrekk, korte høyde, medfødt hjertefeil, problemer med blødninger, og skjelett misdannelser., Ansiktstrekk inkluderer er stor avstand mellom øyne, lyse øyne, lav-sett ører, kort hals, og en liten underkjeve. Hjerte-problemer kan omfatte pulmonal ventilen stenose. Bryst bein kan enten stikke opp eller synke ned, mens ryggen kan være unormalt buet. Intelligens i den syndrom er ofte vanlig. Komplikasjoner av NS-kan inkludere leukemi.
En rekke genetiske mutasjoner kan resultere i Noonan syndrom. Tilstanden kan være arvet fra en persons foreldre som en autosomal dominant tilstand eller oppstå som ny mutasjon., Noonan syndrom er en type RASopathy, er den underliggende mekanismen som innebærer overactivation innenfor RAS/MAPK celle signaliserer veien. Diagnosen kan være mistenkt basert på symptomer, bildediagnostikk og blodprøver. Bekreftelse kan oppnås med genetisk testing.
Ingen kur for NS er kjent. Behandlingen er basert på symptomer og underliggende problemer, og ekstra støtte i skolen kan være nødvendig. Vekst hormone terapi i løpet av barndommen kan øke en berørt person endelige høyde. Langsiktige resultater vanligvis avhenge av alvorlighetsgraden av hjerteproblemer.,
anslagsvis 1 av 1000 personer er mildt rammet av NS, mens om lag 1 i 2,000 har en mer alvorlig form av sykdommen. Hannene ser ut til å være påvirket oftere enn kvinner. Tilstanden ble først beskrevet i 1883 og ble oppkalt etter Amerikansk pediatrisk kardiolog Jacqueline Noonan, som beskrevet tilfeller i 1963.
Tegn og symptomer

Unormal funksjoner av Noonan syndrom i en alder av 3 måneder: Merk øyenbryn skrå og venstre øyelokket slippe.,

Unormal funksjoner av Noonan syndrom i en alder av 3 måneder: Merk lav-set, posteriorly roteres, og unormalt dannet øret.
De mest vanlige tegn som fører til diagnostisering av Noonan syndrom er unike ansiktskjennetegn og muskel-funksjoner. Ansikts egenskaper er mest fremtredende i barndommen, blir mindre tydelig med alderen hos mange mennesker med Noonan syndrom.,
Head
Noen av de karakteristiske trekk ved Noonan syndrom inkluderer et stort hode med overflødig hud på baksiden av halsen, lav hårlinje på nakke av halsen, høye hårfestet på forsiden av hodet, trekantet ansikt form, bred panne, og en kort, svømmehud halsen.
I øynene, hypertelorism (allment sett øynene) er et avgjørende kjennetegn, til stede i 95% av mennesker med Noonan syndrom., Dette kan være ledsaget av epicanthal folder (ekstra fold i huden på det indre hjørnet av øyet), ptose (drooping av øyelokk), proptosis (svulmende øyne), strabismus (innover eller utover for å slå av øynene), nystagmus (krampetrekninger bevegelse av øynene) og refraktiv visuelle feil.
nesen kan være små, brede, og snudd på hodet.
Utvikling av munnen, kan også bli påvirket i Noonan syndrom., Dette kan resultere i dypt rillet philtrum (overleppa linje) (over 90%), micrognathia (underdimensjonert underkjeve), høye buede ganen, artikulasjon, vanskeligheter (tenner ikke på linje), noe som kan føre til dental problemer. Lik den muskuløse manifestasjoner ovenfor, er det i munn, dårlig tunge kontroll kan observeres.,
Huden
Huden tegn og symptomer i Noonan syndrom inkluderer lymphedema (lymfe hevelse i ekstremiteter), keloid dannelse, overdreven arr-dannelse, hyperkeratosis (overdevelopment av ytre hud lag), pigmentert nevi (mørk pigmentert hud flekker), – og bindevev sykdom
Muskel
Misdannelser i beina og ekstremiteter kan oppstå i Noonan syndrom. Dette kan manifestere seg som uten omsvøp endte fingre, ekstra polstring på fingre og tær, ødem på baksiden av hendene og topper av føtter, og cubitus valgus (bredt bærer vinkel i albuene).,
For kortvokste, veksthormon blir noen ganger kombinert med IGF-1 (eller som et alternativ, IGF-1 som en stand-alone) kan brukes for å oppnå en økt høyde/endelige høyde raskere. Den endelige voksne høyde av personer med Noonan syndrom er om 161-167 cm hos menn og 150-155 cm hos kvinner, som nærmer seg den nedre grensen for det normale.
Spinale misdannelser kan være tilstede opp til 30% av tiden, og dette kan kreve kirurgi for å rette opp i over 60% av disse tilfellene., Andre muskel-og manifestasjoner i Noonan syndrom er assosiert med udifferensierte omkringliggende vev lidelser som kan være forbundet med felles kontrakturer (tetthet) eller hypermobilitet (løst). Flere faktorer kan presentere i form av winging av scapula, skoliose, bryst, bein primærfaktor (pectus carinatum), bryst, bein depresjon (pectus excavatum). Muskel misdannelser kan presentere som hypotoni (lav muskeltonus), som kan føre til lordosis (økt hul i ryggen) på grunn av dårlig abdominal muskel tone.,
Hjerte
Noonan syndrom er den nest mest vanlige syndromic årsak til medfødt hjertefeil. Dette inkluderer pulmonal valvulær stenose (50-60%), atrial septal feil (10-25%), ventrikulære septal feil (5-20%) og hypertrofisk kardiomyopati (12-35%).
Lungene
Restriktive lungefunksjonen har blitt rapportert i noen mennesker.
Fordøyelsessystemet
En rekke ulike gastrointestinal (GI) symptomene har vært forbundet med Noonan syndrom., Disse inkluderer svelgevansker, lave gut motilitet, gastroparese (forsinket gastrisk tømming), intestinal malrotation, og hyppig eller kraftig oppkast. Disse fordøyelsesenzymer problemer kan føre til redusert appetitt, til mistrivsel fra barndom til pubertet (75%), og av og til behovet for et materør.{ Det har vært assosiert med genet for Von Willebrand disease, Amegakaryocytic trombocytopeni (lavt antall blodplater), forlenget aktivert partiell thromboplastin tid, kombinert koagulering feil., Når de er til stede, disse Noonan-syndrom tilhørende lidelser kan være forbundet med en predisposisjon for lett blåmerker eller blødning.
Nevrologiske
noen Ganger, Chiari malformasjon (type 1),kan forekomme, noe som kan føre til hydrocephalus. Anfallene har også vært rapportert.
Forårsaker

NS er vanligvis arvelig i en autosomal dominant mønster med variabel uttrykk.,
Tilbakefall i søsken og tilsynelatende overføring fra foreldre til barn har lenge foreslått en genetisk feil med autosomal dominant arv og variable uttrykk. Mutasjoner i Ras/mitogen-aktivert protein kinase signaliserer trasé er kjent for å være ansvarlig for omtrent 70% av NS tilfeller.
Personer med NS har opp til en 50% sjanse for å overføre det til sine avkom., Det faktum at et berørte foreldre er ikke alltid identifisert for barn med NS foreslår flere muligheter:
- Manifestasjoner kan være så diskré som å gå ukjente (variabel expressivity)
- NS er sammensatt gruppe, bestående av mer enn en lignende tilstand av ulike årsaker, og noen av disse kan ikke være arvelig.
- En høy andel av tilfellene kan representere nye, sporadiske mutasjoner.,
| Type | Online Mendelian Inheritance in Man database | – Genet | År funnet | Locus | % i tilfeller | Beskrivelse | Refs. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | Den PTPN11 genet koder protein tyrosin fosfatase SHP-2., Dette proteinet er en komponent av flere intracellulære signal transduksjon trasé involvert i embryonale utvikling som modulerer celledeling, differensiering, og migrasjon, inkludert en mediert av epidermal growth factor receptor, som er viktig i dannelsen av semilunar hjertet ventiler. Duplisering av kromosom regionen inneholder PTPN11 kan også resultere i NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
Heterozygote mutasjoner i NRAS, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, og CBL har også vært forbundet med en mindre andel av NS og relaterte phenotypes.
En tilstand kjent som «nevrofibromatose-Noonan syndrom» er forbundet med neurofibromin.
Diagnose
NS kan være genetisk bekreftet av tilstedeværelsen av noen av de kjente mutasjoner som er nevnt ovenfor., Imidlertid, til tross for identifisering av 14 utløsende gener, fravær av en kjent mutasjon vil ikke utelukke diagnosen, som mer, som-ennå uoppdagede gener kan føre til NS. Dermed, diagnostisering av NS er fortsatt basert på kliniske funksjoner. Med andre ord, det er gjort når en lege mener at en person har nok funksjoner til å rettferdiggjøre etiketten. Den viktigste verdier til å gjøre en genetisk diagnose er at det guider ytterligere medisinsk og utviklingsmessige evalueringer, det utelukker andre mulige forklaringer på funksjoner, og det gir mer nøyaktige gjentakelsesrisiko estimater., Med mer genotype-fenotype korrelasjon studier har blitt utført, en positiv genetisk diagnose vil hjelpe legen å være klar over mulige uregelmessigheter som er spesifikke for det enkelte gen mutasjon. For eksempel, en økning i hypertrofisk kardiomyopati er sett i folk med en mutasjon i KRAS og en økt risiko for juvenil progenitorceller leukemi finnes for en mutasjon av PTPN11. I fremtidige studier kan det føre til en målrettet forvaltning av NS symptomer som avhenger av hva genetisk mutasjon en person har.,
Før fødselen
Prenatal funksjoner som kan føre leger til å vurdere en diagnose av Noonan syndrom inkluderer cystisk hygroma, økt nuchal gjennomskinnelighet, pleural effusjon, og ødem.
differensialdiagnose
Mens Turner syndrom har likheter med nedsatt avvik og utviklingsmål forsinkelse, Turner syndrom er bare funnet i kvinner og uttrykker ofte annerledes. I Turner syndrom, det er en lavere forekomst av utviklingsmessige forsinkelser, venstre-sidig hjertet feil er konstant og forekomsten av nedsatt misdannelser er mye lavere.,
Andre RASopathies
- Watson syndrom – Watson Syndrom har en rekke lignende egenskaper med Noonan Syndrom som kortvoksthet, pulmonal ventilen stenose, variabel intellektuelle utvikling, og huden pigment forandringer.
- Cardiofaciocutaneous (CFC) – syndromet- CFC-syndrom er svært lik Noonan Syndrom på grunn av lignende hjerte-og lymfesystemet funksjoner. Imidlertid, I CFC-syndrom intellektuell funksjonshemning og gastrointestinale problemer er ofte mer alvorlig og uttalt.,
- Costello syndrom – Som CFC-syndrom, Costello syndrom har overlappende funksjoner med Noonan Syndrom. Men forholdene kan være preget av deres genetiske årsak.
- Nevrofibromatose 1 (NF1)
- Williams syndrom
Management
behandling varierer avhengig av komplikasjoner, men har en tendens til å være ganske standard, og reflekterer behandling av den generelle befolkningen., Retningslinjer for forvaltningen er delt inn etter systemer, inkludert general, utviklingsmessige, tannlege, vekst og fôring, kretsløpssystem, nødvendige audiologiske, haematological, nyre og skjelett, som står for tiltak som skal iverksettes ved diagnose, etter diagnose og hvis symptomatisk, har blitt publisert av en Amerikansk konsortium.
Spesielt, behandling av kardiovaskulære komplikasjoner ligner som av den generelle befolkningen og behandling av blødning diathesis er ledet av bestemte faktor mangel eller aggregering.,
- Nevropsykologisk testing anbefales for å finne styrker og utfordringer til å tilpasse støtten som trengs for skole og karriere.
- Pedagogisk tilpasning som en individualisert utdanningsprogram planen er noen ganger nødvendig for school-aged children.
- Tale terapi hvis tale og artikulasjon sakene
- fysioterapi og ergoterapi for grov – og fin-motor forsinkelser
- Hypotoni og motoriske vansker ofte innvirkning håndskrift. Hotellrom for å minske håndskrift krav vil forbedre ytelsen og spare langsiktig håndfunksjon.,
- Periodiske følge opp og livslang oppfølging av avvik som er funnet i et system, spesielt hjerte-karsystemet, anbefales.
Anestesi risiko
Selv om noen mennesker med Noonan syndrom har blitt rapportert å utvikle malign hypertermi, genet mutasjon av sykdommer som er kjent for å være assosiert med malign hypertermi er annerledes enn Noonan syndrom.,
Prognose
levetiden av personer med Noonan syndrom kan være lik den generelle befolkningen, men Noonan syndrom kan være assosiert med flere helsemessige forhold som kan bidra til dødelighet. Den største bidragsyter til dødelighet hos personer med Noonan syndrom er komplikasjoner av hjerte-og karsykdommer. Prognosen er derfor i stor grad avhengig av tilstedeværelse eller fravær av hjerte-og karsykdommer, samt type og alvorlighetsgrad av sykdommen (hvis sykdommen er til stede)., Mest merkbart er Noonan syndrom med hypertrofisk kardiomyopati er assosiert med økt dødelighet.
Logg på
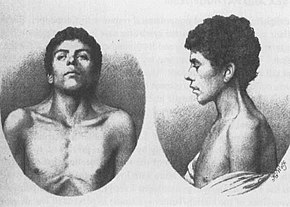
De eldste kjente tilfellet av NS, beskrevet i 1883 av Kobylinski
Jacqueline Noonan var å praktisere som en pediatrisk kardiolog ved University of Iowa da hun la merke til at barn med en sjelden type hjertefeil, valvulær pulmonal stenose, hadde ofte et karakteristisk utseende, med kortvoksthet, svømmehud nakke, bred avstand mellom øyne, og lav-sett ører., Både gutter og jenter, ble berørt. Disse egenskapene var noen ganger sett kjøre i familier, men var ikke assosiert med brutto kromosomavvik. Hun studerte 833 personer med Noonan syndrom ved medfødte hjertefeil klinikk, på jakt etter andre medfødte misdannelser, og i 1963 presenterte et paper: «Forbundet ikke-kardial misdannelser hos barn med medfødt hjertefeil». Dette er beskrevet 9 barn som i tillegg til medfødte hjertefeil hadde karakteristiske ansiktstrekk, brystet deformiteter og kortvoksthet.
Dr. Johannes Opitz, en tidligere student av Dr., Noonan-tallet, først begynte å kalle tilstanden «Noonan syndrom» når han så barn som så ut som de som Dr. Noonan hadde beskrevet. Dr. Noonan produsert en artikkel med tittelen «Hypertelorism med Turner Phenotype» i 1968, hvor hun studerte 19 pasienter som har vist symptomer som tyder på Noonan Syndrom. I 1971 på Symposium av Hjerte-feil, navnet ‘Noonan syndrom» ble offisielt anerkjent.
















Legg igjen en kommentar