Innledning
Intellectual disability (ID) påvirker nesten 1-2% av befolkningen og er den vanligste nevrologiske lidelse (NDD). Et betydelig antall av ID-pasienter er funnet å ha en genetisk årsak (anmeldt i Bessa et al., 2012)., Genome-wide analyse teknikker som benyttes til undersøkelse av etiologi ofte føre til identifisering av svært sjeldne nesten privat varianter, innsamling av pasienter med endringer i det samme genet være en avgjørende faktor for definisjon av en ny klinisk enhet.
Tidligere i år, pasienter husing mutasjoner i EBF3 genet har blitt beskrevet, presentere en nevrologiske syndrom, inkludert ID, ataksi, hypotoni, mild ansikts dysmorphisms, og genitourinary misdannelser (OMIM 617330) (Chao et al., 2017; Skader et al., 2017; Sleven et al., 2017)., Den EBF3 (Tidlig B-celle Faktor 3) – genet koder for et medlem av den svært bevart tidlig B-celle faktor transkripsjonsfaktor familie, uttrykt ved høye nivåer i utvikling av nervesystemet (data hentet fra GTEx Portal). EBF3 er en transcriptional mål av ARX, og vist seg å være regulert av NeuroD og ARX (Friocourt og Parnavelas, 2011). ARX blir en transkripsjonsfaktor kritisk for embryonale utvikling som, for mange år, har vært forbundet med et bredt spekter av nevrologiske sykdommer., Intellektuell svekkelse, sentralnervesystemet og genitourinary avvik observert i pasienten med både mutasjoner i EBF3 og ARX kan reflektere bidrag av både proteiner til samme molekylære og cellulære prosesser (Chao et al., 2017). EBF3 funksjonen har også blitt studert i dyremodeller. Ablasjon av sin orthologs i worms og flyr fører til svekkelse av nevronal utvikling (Prasad et al., 1998; Hattori et al., 2013)., I mus, banket ut Ebf3 fører til neonatal dødelighet og neuronal migrasjon feil, med svikt i olfactory nevroner prosjekt for å dorsal olfactory bulb (Wang et al., 2004). Den eksakte patogene mekanismer for EBF3 mutasjoner er ennå ikke fullt ut belyst, men den type varianter som er beskrevet så langt tyder på at haploinsufficiency, få av funksjon, og dominerende negative er mulige patogene mekanismer for varianter beskrevet (Chao et al., 2017; Sleven et al., 2017).,
I dette arbeidet bidrar vi med en pasient med den minste sletting (600 Kb) rapportert til dato som påvirker helheten i EBF3 genet, og med en klinisk presentasjon overlappende at pasienter med EBF3 enkelt nukleotid varianter (SNVs). I tillegg ønsker vi å gjøre en klinisk sammenligning av pasienter med tidligere publiserte stor terminal 10q slettinger og rapport at det, til tross for forskjeller i størrelse, det er en betydelig fenotypiske overlapp mellom pasienter med disse endringer., Disse funnene legg til i nåværende kunnskap, og for EBF3 relaterte lidelser og støtte EBF3 haploinsufficiency som sentrale i den nevrologiske syndrom forbundet med 10qter slettinger.
Materialer og Metoder
pasienten ble konstatert i en stor studie av nevrologiske lidelser, i Portugal, der registrering av pasienter og familier ble gjort av henvisende lege, klinisk informasjon var samlet i en anonym database i henhold til portugisisk Data Protection Authority (CNPD) og skriftlig informert samtykke ble innhentet for alle deltakerne., Informert samtykke til å presentere pasienten ble levert av moren til genetisk undersøkelse og publisering av resultater (inkludert bilder). Studien ble godkjent av etisk komité ved Senter for Medisinsk Genetikk Dr. Jacinto Magalhães, National Health Institute Dr. Ricardo Jorge.
Genomisk DNA ble ekstrahert fra perifert blod ved hjelp av enten Citogene® DNA isolert sett (Citomed, Portugal). aCGH ble utført ved bruk av Agilent 180 K array (AMADID:023363) mot en diploid DNA-referanse (Kreatech er MegaPoll Referanse DNA, Kreatech Diagnostikk, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., Analysen ble gjennomført i et 7500-RASK Real-Time PCR-maskin (Applied Biosystems, Foster City, CA, USA) ved hjelp av Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) i henhold til produsentens anbefalinger og i henhold til de generelle retningslinjene for qPCR. Spesifisiteten av hver reaksjon ble bekreftet ved generering av en smelte kurve for hver av de forsterkede fragmenter. Primeren effektivitet ble beregnet ved generering av en standard kurve montering akseptert normal effektivitet prosent (primere brukt oppført i Supplerende Data)., Ct-verdier oppnådd for hver test ble analysert i DataAssist™ – programvare (Applied Biosystems, Foster City, CA, USA).
Resultater
Her beskriver vi en pasient med en de novo sletting påvirker EBF3. Pasienten er en 11 år gammel jente med alvorlige ID (global utvikling kvotient = 27 på 7 år), født fra ikke-consanguineous foreldre og med ingen familie historie av nevrologiske sykdommer. Hun ble født etter en biamniotic bichorionic twin graviditet (hennes søster være sunn), ved vaginal levering, 35 uker av svangerskapet. Fødselen var parametrene: vekt, 1,830 g (P3); lengde, 42.,5 cm (P10), og OFC, 30.6 cm (P10), med en Apgar score på 8/9 (1. og 5. min., henholdsvis). Den neonatale periode var komplisert med sepsis og diagnostikk av arvelige spherocytosis (arvet fra sin mor). Globale utviklingsmål forsinkelse ble nevnt i den første måneder, med hodet kontroll oppnås ved 12 måneder, sitter ved 18 måneder, uavhengig gå på 30 måneder, og ingen ord talt i en alder av 3 år., Hun hadde pyelonefritt på 19 måneder (renal ultralyd viste ingen avvik), gastroøsofageal refluks og residiverende otitis media, med mekanisk hørselstap som kreves kirurgiske inngrep og et høreapparat. Epilepsi var mistenkt i 5 måneder (episoder av suspendert aktivitet), men EEG var normal.
Hun ble først observert i en alder av 3 år, 5 måneder (Figur 1A), B), hvor hun viste muskler hypotoni, hypoton ansikt, strabismus, og redusert følsomhet for smerte., Hun har også presentert mild dysmorphic funksjoner (Figur 1A): trekantet ansikt, små lav-sett ører med fremtredende anti-helix, buede øyebryn, anteverted nares, bulbous nese tips, liten munn med ned slått hjørnespark, spiss hake, kort hals, og fremtredende finger fosterets pads, samt en mild kortvoksthet (89 cm, tilsvarende rundt 2SD).

Figur 1. (En) Ansikts utseende av pasienten på 3 år og 5 måneder som viser liten og lav-sett ører med fremtredende anti-helix og (B) fetal pads i fingrene., (C) Ansikts utseende av pasienten ved 11 års alder. (D) Markert i grått 600 Kb sletting i 10q26.3-regionen, en zoom inn på EBF3 gen i DGV database avslører eksistensen av 3 slettinger i 3-kontroller som påvirker de første 6 exons av EBF3 (NM_001005463); CNVs innenfor denne regionen finnes i kontroll bestander inkluderer slettinger nvs825626 (til stede i 1/31 individer), nvs552315 (til stede i 1/17421 individer) og nsv552316 (til stede i 1/31 enkeltpersoner). (E) skjematisk fremstilling av EBF3 transkripsjoner.,
Brain MR ble utført på 6 år, men ingen avvik ble notert. I en alder av 10 år ble hun revurderes; hun hadde fortsatt residiverende otitis media, men ellers var i god global helse. Språket var svært dårlig (to ord setninger som snakkes etter 8 år). Hun hadde atferdsproblemer, med stereotypic bevegelser (roterende bevegelser, tygge på klær, leder retropulsion), scoring for alvorlig autism spectrum disorder (ADI-R og ADOS) på 7 år; hun viste uro og aggressiv atferd (auto og hetero) og ble medisinert med antipsykotiske legemidler., En ortopedisk kirurgi ble utført for pes planus. Ansiktstrekkene var lik de som er beskrevet tidligere, med linjeavstand øvre sentrale fortenner (Figur 1C); hun hadde briller for strabismus og hypermetropia.
Analyse av genomisk DNA ved aCGH åpenbart en de novo 600 kb sletting i 10p26.3 (Figur 1D) som påvirker tre gener—MGMT (koding enzymet O-6-methylguanine-DNA-metyltransferase, som er involvert i DNA-reparasjon), EBF3 og GLRX (koding glutaredoxin, en liten thioltransferase som fjerner protein GSH adducts), som EBF3 var mest sannsynlig sykdomsassosierte gener.,
Diskusjon
presentert pasienten ble først analysert ved aCGH for noen år siden. På den tiden av aCGH-analyse, eksistensen av de tre variantene finnes i Databasen av Genomisk Varianter (DGV)1 som påvirker de fem første exons av EBF3 genet (Figur 1E, Parker et al., 2010; Cooper et al., 2011), så vel som fravær av andre kjente sykdomsfremkallende mutasjoner i dette genet, lede oss til å klassifisere det som en variant av ukjent betydning (VOUS). Imidlertid, den nye publikasjoner av EBF3 mutasjoner (Chao et al., 2017; Skader et al., 2017; Sleven et al.,, 2017) og den kliniske likheter med den rapporterte tilfeller, føre oss til å re-vurdere-varianten, og få oss til å tro at EBF3 sletting kan faktisk være regnskap for sykdom hos pasienten. En av de aspektene som er reist tvil om pathogenicity av denne varianten i første omgang var eksistensen av befolkningen kontrollerer lager slettinger av de første seks exons av dette genet, i heterozygosity (data hentet fra DGV database som i februar 2017) (Figur 1D). Selv om en utskrift av EBF3 starter i Exon13b er oppført i Ensembl database (ENST00000440978.,1) (Figur 1A), som kan forklare hvordan sletting av den første exons kan etter hvert resultere i en normal fenotype, denne protokoll utelukker DNA-bindende domene av EBF3, og dens uttrykk og funksjonelle relevans har ikke vært preget. Ved revurdering, men CNVs beskrevet av Cooper og kolleger (nsv552315 og nsv552316) (Chao et al., 2017; Skader et al., 2017; Sleven et al.,, 2017) ble vurdert til å være på terskelen til påvisning av SNP microarray, og kan ikke være grunnlag for utelukkelse av en kandidat gen, særlig i lys av den sterke genetiske og funksjonelle bevis for relevansen av EBF3 mutasjoner til sykdom (Evan Eichler, Greg Bradley Cooper og Coe, personlig kommunikasjon).,
Vår pasient viser mange kliniske likheter med tidligere beskrevet pasienter med mutasjoner i EBF3 (21 tilfeller er oppsummert i Tabell 1), for eksempel globale utviklingsmål forsinkelse, forsinket uttrykksfulle tale, hypotoni, økt smerteterskel, atferdsproblemer og karakteristiske ansiktstrekk (lang/trekantet ansikt, stor panne, hypoton ansiktet)., Imidlertid, selv om vår pasient hadde betydelig forsinkelse i motorisk utvikling, ingen signifikante ataksi ble oppdaget (med noen begrensninger i klinisk undersøkelse, som barnet ikke var samarbeidsvillig), og ingen lillehjernen avvik var til stede i hjernen MR-undersøkelse.

Tabell 1. Kliniske sammenligning av saken med de rapporterte tilfellene med punkt mutasjoner/indels i EBF3 genet.
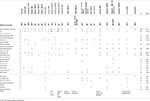
Tabell 2., Kliniske sammenligning av saken med de rapporterte tilfellene med slettinger som påvirker 10q26 cytoband.
EBF3 varianter beskrevet i litteraturen i begynnelsen av 2017 inkluderer punkt mutasjoner spådd å være skadelige og små innsettinger og slettinger som fører til ramme sletting av nøkkel aminoacids eller til en frameshift, forutsigbart forårsaker tidlig trunkering av den resulterende protein eller tull-mediert forfall., Den mutasjoner var konsentrert på deler av genet koding av DNA-bindende domene av EBF3, og ble spådd gjennom ulike metoder for å føre til et tap av funksjon av denne transkripsjonsfaktor, og dermed noe som tyder på redusert funksjon og haploinsufficiency som mekanisme underliggende nevrologiske forstyrrelser i disse pasientene. Knock-out mus for Ebf3 er beskrevet til å presentere neonatal dødelighet og neuronal migrasjon feil, med svikt i olfactory nevroner til prosjekt til dorsal olfactory bulb (Wang et al.,, 2004), men ingen beskrivelse er laget av en fenotypen i heterozygot dyr, som faktisk er presentert som styrer i mange av forsøkene, og dermed ikke støtte haploinsufficiency modell. Vi gjorde forsøk på å få tak i og studere den nevrologiske fenotypen av disse dyrene, men var ikke vellykket, som Ebf3(O/E2) knock-out linje kan ha blitt avviklet (Joseph W. Lewcock, personlig kommunikasjon). Imidlertid, gjeldende sak sammen med pasienter som er oppsummert i Tabell 2, støtter hypotesen om EBF3 haploinsufficiency som forårsaker sykdom.,
I sammendraget, gjeldende beskrivelse forsterker EBF3 tap av funksjon/haploinsufficiency som en årsak til nevrologiske sykdom, og forsterker foreningen av dette genet med en karakteristisk klinisk syndrom innenfor dette spekteret.
Forfatter Bidrag
FL utført molekylære studier og analysert molekylære data. MG og GS samlet kliniske data. FL, JP, og PM gjennomgått alle EBF3 mutasjon tilfeller i litteraturen. FL, GS, JP, og PM skrevet på papir. PM fått finansiering for denne studien. Studien ble utført under ledelse av PM.,
Midler
FCT—Fundação para en Ciência e en Tecnologia i prosjekter og stipender (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Denne artikkelen har blitt utviklet under prosjektets omfang NORTE-01-0145-FEDER-000013, støttes av den Nordlige Portugal Regionale Program (NORTE 2020), under Portugal 2020 Partnerskap Avtale, gjennom det Europeiske Regionale utviklingsfondet (FEDER).
interessekonflikt Uttalelse
JP var ansatt i selskapet CGC Genetikk.,
Den andre forfattere erklærer at forskningen ble utført i fravær av kommersielle eller finansielle forhold som kan oppfattes som en potensiell interessekonflikt.
Erkjennelsene
Vi vil gjerne takke pasienten og hennes familie for deltakelse i den genetiske studier og for å la denne publikasjonen. Vi er takknemlige for Dr. Ana Maria Fortuna og Dr. Margarida Reis i Lima for å gjøre dette prosjektet mulig, og ved å legge til rette vårt samarbeid med CGM.
Fotnoter
1., ^Database av Genomisk Varianter Tilgjengelig på: http://dgv.tcag.ca/dgv/app/home .
Friocourt, G., og Parnavelas, J. (2011). Identifisering av Arx mål lanserer nye kandidater til styre kortikale interneuron migrasjon og differensiering. Front. Celle. Neurosci. 5:28. doi: 10.3389/fncel.2011.00028
PubMed Abstrakt | CrossRef Full Tekst | Google Scholar
















Legg igjen en kommentar