Einführung
Intellektuelle Behinderung (ID) betrifft fast 1-2% der Bevölkerung und ist die häufigste neurodevelopmental Disorder (NDD). Eine beträchtliche Anzahl von ID-Patienten hat eine genetische Ursache (überprüft in Bessa et al., 2012)., Genomweite Analysetechniken, die derzeit zur Untersuchung der Ätiologie verwendet werden, führen häufig zur Identifizierung sehr seltener, fast privater Varianten, wobei die Sammlung von Patienten mit Veränderungen desselben Gens ein entscheidender Aspekt bei der Definition einer neuen klinischen Entität ist.
Anfang dieses Jahres wurden Patienten beschrieben, die Mutationen im EBF3-Gen aufweisen und ein Neuroentwicklungssyndrom aufweisen, einschließlich ID, Ataxie, Hypotonie, leichten Gesichtsdysmorphismen und Urogenitalanomalien (OMIM 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., Das EBF3-Gen (Early B cell Factor 3) kodiert für ein Mitglied der hochkonservierten Transkriptionsfaktorfamilie der frühen B-Zell-Faktoren, die auf hohem Niveau im sich entwickelnden Nervensystem exprimiert wird (Daten aus dem GTEx-Portal). EBF3 ist ein Transkriptionsziel von ARX und wird nachweislich durch NeuroD und ARX reguliert (Friocourt und Parnavelas, 2011). ARX kodiert einen für die Embryonalentwicklung kritischen Transkriptionsfaktor, der seit vielen Jahren mit einer Vielzahl von neurologischen Entwicklungsstörungen in Verbindung gebracht wird., Die intellektuelle Beeinträchtigung, das zentrale Nervensystem und Urogenitalanomalien, die bei Patienten mit beiden Mutationen in EBF3 und ARX beobachtet wurden, könnten den Beitrag beider Proteine zu denselben molekularen und zellulären Prozessen widerspiegeln (Chao et al., 2017). EBF3-Funktion wurde auch in Tiermodellen untersucht. Die Ablation seiner Orthologen bei Würmern und Fliegen führt zu einer Beeinträchtigung der neuronalen Entwicklung (Prasad et al., 1998; Hattori et al., 2013)., Bei Mäusen führt das Ausschlagen von Ebf3 zu neonataler Letalität und neuronalen Migrationsdefekten, wobei das Versagen von Geruchsneuronen auf den dorsalen Geruchszwiebel projiziert wird (Wang et al., 2004). Die genauen pathogenen Mechanismen von EBF3-Mutationen sind noch nicht vollständig aufgeklärt, aber die bisher beschriebenen Varianten legen nahe, dass Haploinsuffizienz, Funktionsgewinn und dominantes Negativ mögliche pathogene Mechanismen für die beschriebenen Varianten sind (Chao et al., 2017; Sleven et al., 2017).,
In dieser Arbeit tragen wir mit einem Patienten mit der kleinsten Löschung (600 Kb) – Datum gemeldet Auswirkungen auf die Gesamtheit von EBF3-gen und mit einer klinischen Präsentation überlagerung, die von Patienten mit EBF3 Einzel-Nukleotid-Varianten (SNVs). Darüber hinaus führen wir einen klinischen Vergleich der Patienten mit zuvor veröffentlichten großen terminalen 10q-Deletionen durch und berichten, dass trotz der Größenunterschiede eine signifikante phänotypische Überlappung zwischen Patienten mit diesen Veränderungen besteht., Diese Ergebnisse ergänzen das aktuelle Wissen über EBF3-bedingte Störungen und unterstützen EBF3-Haploinsuffizienz als Schlüssel für das Neuroentwicklungssyndrom im Zusammenhang mit 10qter-Deletionen.
Materialien und Methoden
Der Patient wurde im Rahmen einer großen Studie zu neurologischen Entwicklungsstörungen in Portugal ermittelt, in der die Registrierung der Patienten und Familien vom überweisenden Arzt durchgeführt wurde, Klinische Informationen wurden in einer anonymen Datenbank gemäß der portugiesischen Datenschutzbehörde (CNPD) gesammelt und eine schriftliche Einwilligung in Kenntnis gesetzt wurde für alle Teilnehmer eingeholt., Die Einverständniserklärung für den vorliegenden Patienten wurde von der Mutter für die genetische Untersuchung und Veröffentlichung der Ergebnisse (einschließlich Fotos) erteilt. Die Studie wurde von der Ethikkommission des Zentrums für medizinische Genetik Dr. Jacinto Magalhães, National Health Institute Dr. Ricardo Jorge genehmigt.
Genomische DNA wurde entweder mit Citogene® DNA isolation Kit (Citomed, Portugal) aus peripherem Blut extrahiert. aCGH wurde mittels Agilent 180 K Array (AMADID:023363) gegen eine diploide DNA Referenz (Kreatech ‚ s MegaPoll Reference DNA, Kreatech Diagnostics, Amsterdam) durchgeführt., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., Die Analyse wurde in einer 7500-SCHNELLEN Echtzeit-PCR-Maschine (Applied Biosystems, Foster City, CA, USA) unter Verwendung von Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) gemäß den Empfehlungen des Herstellers und gemäß den allgemeinen Richtlinien für qPCR durchgeführt. Die Spezifität jeder Reaktion wurde durch die Erzeugung einer Schmelzkurve für jedes der amplifizierten Fragmente verifiziert. Die Primereffizienz wurde durch die Erzeugung einer Standardkurve berechnet, die dem akzeptierten normalen Effizienzprozentsatz entspricht (verwendete Primer in ergänzenden Daten aufgeführt)., Die für jeden Test erhaltenen Ct-Werte wurden in der DataAssist™ – Software (Applied Biosystems, Foster City, CA, USA) analysiert.
Ergebnisse
Hier beschreiben wir einen Patienten mit einer de novo Deletion, die EBF3 betrifft. Die Patientin ist ein 11-jähriges Mädchen mit schwerem ID (globaler Entwicklungsquotient = 27 im Alter von 7 Jahren), das von nicht-konsanguinen Eltern geboren wurde und in der Familiengeschichte keine neurologischen Entwicklungsstörungen aufweist. Sie wurde nach einer biamniotischen bichorionischen Zwillingsschwangerschaft (ihre Schwester ist gesund) durch vaginale Entbindung nach 35 Schwangerschaftswochen geboren. Geburtsparameter waren: Gewicht, 1.830 g (P3); Länge, 42.,5 cm (P10); und in OFC, 30.6 cm (P10), mit einem Apgar-score von 8 auf 9 (1. und 5. min, respectively). Die Neugeborenenperiode war kompliziert mit Sepsis und der Diagnose einer erblichen Sphärozytose (von ihrer Mutter geerbt). In den ersten Monaten wurde eine globale Entwicklungsverzögerung festgestellt, wobei die Kopfkontrolle nach 12 Monaten, das Sitzen nach 18 Monaten, das unabhängige Gehen nach 30 Monaten und keine Worte im Alter von 3 Jahren erreicht wurden., Sie hatte nach 19 Monaten Pyelonephritis (Nierenultraschall zeigte keine Anomalien), gastroösophagealen Reflux und rezidivierende Mittelohrentzündung mit leitfähigem Hörverlust, der einen chirurgischen Eingriff und ein Hörgerät erforderte. Epilepsie wurde nach 5 Monaten vermutet (Episoden suspendierter Aktivität), aber das EEG war normal.
Sie wurde zuerst im Alter von 3 Jahren 5 Monaten beobachtet (Abbildungen 1A,B), zu diesem Zeitpunkt zeigte sie Muskelhypotonie, hypotonisches Gesicht, Strabismus und reduzierte Schmerzempfindlichkeit., Sie präsentierte auch milde dysmorphe Merkmale (Abbildung 1A): dreieckiges Gesicht, kleine niedrig gesetzte Ohren mit hervorstechender Anti-Helix, gewölbte Augenbrauen, antevertierte Nasen, Knollennase, kleiner Mund mit heruntergekommenen Ecken, spitzes Kinn, kurzer Hals und prominente Finger fötale Pads sowie eine milde kleinwüchsige Statur (89 cm, entsprechend etwa 2SD).

Abbildung 1. (A) Gesichtsausdruck des Patienten nach 3 Jahren und 5 Monaten, der die kleinen und niedrig gesetzten Ohren mit prominenter Anti-Helix und (B) fetalen Pads in den Fingern zeigt., (C) Gesichtsausdruck des Patienten im Alter von 11 Jahren. (D) Grau hervorgehoben die 600 Kb Deletion im 10q26.3-Bereich; Ein Vergrößern des EBF3-Gens in der DGV-Datenbank zeigt das Vorhandensein von 3 Deletionen in 3-Kontrollen, die die ersten 6 Exons von EBF3 betreffen (NM_001005463); CNVs in dieser Region, die in Kontrollpopulationen gefunden werden, umfassen Deletionen nvs825626 (vorhanden in 1/31-Individuen), nvs552315 (vorhanden in 1/17421-Individuen) und nsv552316 (vorhanden in 1/31-Individuen). (E) Die schematische Darstellung von EBF3-Transkripten.,
Die MRT des Gehirns wurde nach 6 Jahren durchgeführt, es wurden jedoch keine Anomalien festgestellt. Im Alter von 10 Jahren wurde sie neu bewertet; Sie hatte immer noch wiederkehrende Mittelohrentzündung, war aber ansonsten bei guter globaler Gesundheit. Die Sprache war sehr schlecht (zwei Wortsätze nach 8 Jahren gesprochen). Sie hatte Verhaltensprobleme mit stereotypen Bewegungen (rotierende Bewegungen, Kauen auf Kleidung, Kopfretropulsion) und erzielte mit 7 Jahren eine schwere Autismus-Spektrum-Störung (ADI-R und ADOS); Sie zeigte Erregung und aggressives Verhalten (Auto und Hetero) und wurde mit Antipsychotika behandelt., Eine orthopädische Operation wurde für Pes planus durchgeführt. Die Gesichtszüge ähnelten denen, die zuvor beschrieben wurden, mit abgegrenzten oberen mittleren Schneidezähnen (Abbildung 1C); Sie hatte eine Brille für Strabismus und Hypermetropie.
Die Analyse der genomischen DNA durch aCGH ergab eine de novo 600 kb-Deletion bei 10p26. 3 (Abbildung 1D), die drei Gene—MGMT (Kodierung des Enzyms O-6-Methylguanin-DNA-Methyltransferase, die an der DNA-Reparatur beteiligt ist), EBF3 und GLRX (Kodierung von Glutaredoxin, einer kleinen Thioltransferase, die Protein-GSH-Addukte entfernt), von denen EBF3 das wahrscheinlichste krankheitsassoziierte Gen war.,
Diskussion
Der vorgestellte Patient wurde vor einigen Jahren erstmals von aCGH analysiert. Zum Zeitpunkt der aCGH-Analyse bestand die Existenz der drei in der Datenbank der genomischen Varianten (DGV)1 vorhandenen Varianten, die die ersten fünf Exons des EBF3-Gens betrafen (Abbildung 1E; Park et al., 2010; Cooper et al., 2011) sowie das Fehlen anderer bekannter krankheitsverursachender Mutationen in diesem Gen führen dazu, dass wir es als eine Variante unbekannter Bedeutung (VOUS) klassifizieren. Die jüngsten Veröffentlichungen von EBF3-Mutationen (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) und die klinischen Ähnlichkeiten mit den gemeldeten Fällen führen dazu, dass wir die Variante neu bewerten und glauben, dass die EBF3-Deletion tatsächlich für die Krankheit beim Patienten verantwortlich sein kann. Einer der Aspekte, die Zweifel an der Pathogenität dieser Variante aufkommen ließen, war in erster Linie das Vorhandensein von Populationskontrollen mit Löschungen der ersten sechs Exons dieses Gens in Heterozygot (Daten aus der DGV-Datenbank ab Februar 2017) (Abbildung 1D). Auch wenn eine Abschrift von EBF3 ab Exon13b in der Ensembldatenbank aufgeführt ist (ENST00000440978.,1E), die erklären könnte, wie die Deletion der ersten Exons schließlich zu einem normalen Phänotyp führen könnte, dieses Transkript schließt die DNA-Bindungsdomäne von EBF3 aus, und sein Expressionsmuster und seine funktionelle Relevanz wurden nicht charakterisiert. Bei der Neubewertung wurden jedoch die von Cooper und Kollegen beschriebenen CNVs (nsv552315 und nsv552316) (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) als an der Nachweisschwelle durch SNP Microarray und können nicht die Grundlage für den Ausschluss eines Kandidatengens sein, insbesondere angesichts der starken genetischen und funktionellen Evidenz für die Relevanz von EBF3-Mutationen für Krankheiten (Evan Eichler, Greg Cooper und Bradley Coe, persönliche Kommunikation).,
Unser Patient zeigt viele klinische Ähnlichkeiten mit zuvor beschriebenen Patienten mit Mutationen in EBF3 (21 Fälle in Tabelle 1 zusammengefasst), wie globale Entwicklungsverzögerung, verzögerte expressive Sprache, Hypotonie, erhöhte Schmerzschwelle, Verhaltensprobleme und charakteristische Gesichtszüge (langes/dreieckiges Gesicht, große Stirn, hypotonisches Gesicht)., Obwohl unser Patient eine signifikante Verzögerung der motorischen Entwicklung aufwies, wurde keine signifikante Ataxie festgestellt (mit einigen Einschränkungen in der klinischen Untersuchung, da das Kind nicht kooperativ war), und es waren keine Kleinhirnanomalien in der MRT des Gehirns vorhanden.

Tabelle 1. Klinischer Vergleich des vorliegenden Falles mit den gemeldeten Fällen mit Punktmutationen / Indels im EBF3-Gen.
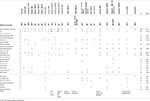
Tabelle 2., Klinischer Vergleich des vorliegenden Falles mit den gemeldeten Fällen mit Löschungen, die das 10q26-Zytoband betreffen.
Die Anfang 2017 in der Literatur beschriebenen EBF3-Varianten umfassen Punktmutationen, von denen vorhergesagt wird, dass sie schädlich sind, und kleine Einfügungen und Löschungen, die zu einer Frame-Deletion von Schlüsselaminsäuren oder zu einem Frameshift führen, was vorhersehbar zu einer frühen Kürzung des resultierenden Proteins oder zu einem Unsinn-vermittelten Zerfall führt., Die Mutationen konzentrierten sich auf Teile des Gens, das für die DNA-Bindungsdomäne von EBF3 kodiert,und wurden durch verschiedene Methoden vorhergesagt, um zu einem Funktionsverlust dieses Transkriptionsfaktors zu führen, was auf eine verminderte Funktion und Haploinsuffizienz als Mechanismus hindeutet, der der neurologischen Entwicklungsstörung bei diesen Patienten zugrunde liegt. Es wird beschrieben, dass Knock-out-Mäuse für Ebf3 neonatale Letalität und neuronale Migrationsdefekte aufweisen, wobei olfaktorische Neuronen nicht in den dorsalen olfaktorischen Bulb projiziert werden können (Wang et al.,, 2004), aber es wird kein Phänotyp bei den heterozygoten Tieren beschrieben, die in vielen Experimenten tatsächlich als Kontrollen dargestellt werden und somit das Haploinsufficiency-Modell nicht unterstützen. Wir bemühten uns, den Neuroentwicklungsphänotyp dieser Tiere zu erhalten und zu untersuchen, waren jedoch nicht erfolgreich, da die Ebf3(O/E2)-Knock-out-Linie möglicherweise eingestellt wurde (Joseph W. Lewcock, persönliche Kommunikation). Der aktuelle Fall unterstützt jedoch zusammen mit den in Tabelle 2 zusammengefassten Patienten die Hypothese einer EBF3-Haploinsuffizienz als krankheitsverursachend.,
In der Zusammenfassung, die aktuelle Beschreibung verstärkt EBF3 Verlust der Funktion/haploinsufficiency als Ursache für neurologische Entwicklungsstörungen, Krankheit und verstärkt die Assoziation dieses Gens mit einem charakteristischen klinischen Syndrom innerhalb dieses Spektrums.
Autorenbeiträge
FL führte die molekularen Studien durch und analysierte die molekularen Daten. MG und GS sammelten klinische Daten. FL, JP und PM überprüften alle EBF3-Mutationsfälle in der Literatur. FL, GS, JP und PM entwarfen das Papier. PM erhielt Mittel für diese Studie. Die Studie wurde unter der Leitung von PM durchgeführt.,
Finanzierung
FCT—Fundação para a Ciência e a Tecnologia innerhalb der Projekte und Stipendien (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Dieser Artikel wurde im Rahmen des Projekts entwickelt.-01-0145- FEDER-000013, unterstützt durch das regionale operationelle Programm Nordportugal (NORTE 2020) im Rahmen des Partnerschaftsabkommens Portugal 2020 über den Europäischen Fonds für regionale Entwicklung (FEDER).
Interessenkonflikterklärung
JP wurde von der Firma CGC Genetics beschäftigt.,
Die anderen Autoren erklären, dass die Forschung ohne kommerzielle oder finanzielle Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagungen
Wir danken der Patientin und ihrer Familie für die Teilnahme an den genetischen Studien und für die Genehmigung dieser Veröffentlichung. Wir sind Dr. Ana Maria Fortuna und Dr. Margarida Reis Lima dankbar, dass sie dieses Projekt ermöglicht und unsere Zusammenarbeit mit CGM erleichtert haben.
Fußnoten
1., ^- Datenbank der Genomischen Varianten Verfügbar unter: http://dgv.tcag.ca/dgv/app/home .
Friocourt, G., und Parnavelas, J. (2011). Identifizierung von Arx-Zielen enthüllt neue Kandidaten zur Kontrolle der kortikalen Interneuron-Migration und-Differenzierung. Front. Zelle. Neurowissenschaften. 5:28. doi: 10.3389/fncel.2011.00028
PubMed Abstract / CrossRef Volltext / Google Scholar
















Schreibe einen Kommentar