Han Turner syndrom, Noonan-Ehmke syndrom, Turner-lignende syndrom, Ullrich-Noonan syndrom
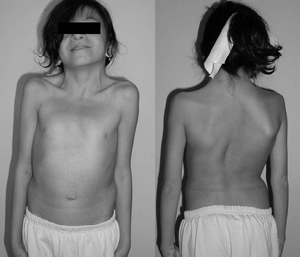
En 12-årig pige med Noonan syndrom. Typisk weebbed hals. Dobbelt strukturel kurve med ribdeformitet.,irmed med genetiske test
Cardiofaciocutaneous syndrom, Turner syndrom, Costello syndrom, neurofibromatosis type 1
Baseret på symptomer
væksthormon
Afhænger af sværhedsgraden af problemer med hjertet
1: 100 (1 i 2.000 alvorlig sygdom)
Noonan syndrom (NS) er en genetisk sygdom, der kan præsentere med mildt usædvanlige ansigtstræk, korte højde, medfødte hjertesygdomme, blødning problemer, og knoglemisdannelser., Ansigtstræk omfatter vidt adskilte øjne, lyse øjne, lavtliggende ører, en kort hals og en lille underkæbe. Hjerteproblemer kan omfatte lungeventilstenose. Brystbenet kan enten stikke ud eller blive nedsænket, mens rygsøjlen kan være unormalt buet. Intelligens i syndromet er ofte normalt. Komplikationer af NS kan omfatte leukæmi.
et antal genetiske mutationer kan resultere i Noonan-syndrom. Tilstanden kan arves fra en persons forældre som en autosomal dominerende tilstand eller forekomme som en ny mutation., Noonan syndrom er en type Rasopati, den underliggende mekanisme, som involverer overaktivering inden for RAS/MAPK-cellesignalvejen. Diagnosen kan mistænkes baseret på symptomer, medicinsk billeddannelse og blodprøver. Bekræftelse kan opnås ved genetisk test.
ingen kur mod NS er kendt. Behandlingen er baseret på symptomer og underliggende problemer, og ekstra støtte i skolen kan være påkrævet. Væksthormonbehandling i barndommen kan øge en berørt persons endelige højde. Langsigtede resultater afhænger typisk af sværhedsgraden af hjerteproblemer.,1 ud af 1000 mennesker er mildt påvirket af NS, mens omkring 1 ud af 2.000 har en mere alvorlig form for tilstanden. Mænd synes at blive påvirket oftere end kvinder. Tilstanden blev først beskrevet i 1883 og blev opkaldt efter den amerikanske pædiatriske kardiolog Jac .ueline Noonan, der beskrev yderligere tilfælde i 1963.
Tegn og symptomer

Unormal funktioner i Noonan syndrom i en alder af 3 måneder: Bemærk den skrå øjenbryn og venstre side af øjenlåget slippe.,

Unormal funktioner i Noonan syndrom i en alder af 3 måneder: Bemærk det lave-sæt, bagtil roteres, og unormalt dannet øre.
de mest almindelige tegn, der fører til diagnosen Noonan syndrom, er unikke ansigtsegenskaber og muskuloskeletale træk. Ansigtsegenskaberne er mest fremtrædende i spædbarnet og bliver mindre tydelige med alderen hos mange mennesker med Noonan-syndrom.,
Chef
Nogle af de karakteristiske træk af Noonan syndrom omfatter et stort hoved med overskydende hud på bagsiden af halsen, lav hårgrænse på nakken af halsen, høj hårgrænse på den forreste del af hovedet, trekantet ansigtsform, bred pande, og en kort, svømmehud hals.
i øjnene er hypertelorisme (bredt set øjne) en definerende egenskab, der er til stede hos 95% af mennesker med Noonan-syndrom., Dette kan være ledsaget af epicanthal folder (ekstra hudfold i inderste øjenkrog), ptose (hængende øjenlåg), proptosis (udstående øjne), strabismus (indad eller udad drejning i øjnene), nystagmus (rykkende bevægelse af øjnene) og brydningsindeks visuelle fejl.
næsen kan være lille, bred og opadvendt.
udvikling af munden kan også påvirkes ved Noonan-syndrom., Dette kan resultere i dybt rillede philtrum (øverste læbe linje) (over 90%), mikrognati (undermålere underkæben), høj buet gane, artikulation vanskeligheder (tænder ikke line up), som kan føre til tandproblemer. I lighed med de muskulære manifestationer ovenfor kan der i munden observeres dårlig tunge kontrol.,
Hud
Hudens tegn og symptomer i Noonan syndrom omfatter lymphedema (lymfeknuder hævelse af ekstremiteter), keloid dannelse, overdreven ardannelse, hyperkeratose (uhæmmet udvikling af ydre hudlag), pigmenterede modermærker (mørkt pigmenteret hud pletter), og bindevævssygdom
Muskel –
Abnormiteter i lemmer og ekstremiteter kan forekomme i Noonan syndrom. Dette kan manifestere sig som ligeud endte fingre, ekstra polstring på fingre og tæer, ødem af bagsiden af hænder og toppe af fødder, og albueben valgus (bred regnskabsmæssig vinkel på albuerne).,
For korte statur, væksthormon er undertiden kombineret med IGF-1 (eller som et alternativ, IGF-1, som en stand-alone) kan bruges til at opnå en øget højde/endelige højde hurtigere. 161-167 cm hos mænd og 150-155 cm hos kvinder, som nærmer sig den nedre grænse for normal.
Spinal abnormiteter kan være til stede op til 30% af tiden, og dette kan kræve operation for at korrigere i over 60% af disse tilfælde., Andre muskuloskeletale manifestationer ved Noonan-syndrom er forbundet med udifferentierede bindevævssygdomme, der kan være forbundet med ledkontrakturer (tæthed) eller ledhypermobilitet (løshed). Yderligere faktorer kan forekomme i form af vinging af scapulaen, skoliose, prominens for brystbenet (pectus carinatum), brystbensdepression (pectus e .cavatum). Muskel abnormiteter kan præsentere som hypotoni (lav muskeltonus), hvilket kan føre til lordose (øget hul i ryggen) på grund af dårlig abdominal muskeltonus.,
hjerte
Noonan syndrom er den næst mest almindelige syndromiske årsag til medfødt hjertesygdom. Dette inkluderer pulmonal valvulær stenose (50-60%), atriale septumdefekter (10-25%), ventrikulære septumdefekter (5-20%) og hypertrofisk kardiomyopati (12-35%).
lunger
restriktiv lungefunktion er rapporteret hos nogle mennesker.
Gastrointestinal
en række forskellige gastrointestinale (GI) symptomer er blevet forbundet med Noonan syndrom., Disse omfatter synke vanskeligheder, lav tarm motilitet, gastroparesis (forsinket gastrisk tømning), tarm malrotation, og hyppig eller kraftig opkastning. Disse fordøjelsesproblemer kan føre til nedsat appetit, manglende trivsel fra spædbarn til pubertet (75%) og lejlighedsvis behovet for et foderrør.{ Det har været forbundet med Von Vonillebrands sygdom, amegakaryocytisk trombocytopeni (lavt antal blodplader), forlænget aktiveret partiel thromboplastintid, kombinerede koagulationsdefekter., Når de er til stede, kan disse Noonan-syndrom ledsagende lidelser være forbundet med en disponering for let blå mærker eller blødning.
neurologisk
lejlighedsvis kan Chiari-misdannelse (type 1) forekomme, hvilket kan føre til hydrocephalus. Der er også rapporteret anfald.
Årsag

NS er typisk nedarvet i en autosomal dominant mønster med varierende udtryk.,
gentagelse hos søskende og tilsyneladende transmission fra forælder til barn har længe antydet en genetisk defekt med autosomal dominerende arv og variabel ekspression. Mutationer i Ras / mitogenaktiverede proteinkinase-signalveje vides at være ansvarlige for omkring 70% af NS-tilfælde.
personer med NS har op til 50% chance for at overføre det til deres afkom., Det faktum, at en af de berørte forældre er ikke altid, der er identificeret for børn med NS antyder, er der flere muligheder:
- Manifestationer kan være så subtile som at gå ukendt (variabel ekspressivitet)
- NS er heterogene, og som omfatter mere end en lignende betingelse af forskellige årsager, og nogle af disse ikke kan være arvelig.
- en høj andel af tilfælde kan repræsentere nye sporadiske mutationer.,
| Type | Online Mendelian Inheritance in Man-database | Gen | År fundet | Locus | % i tilfælde | Beskrivelse | Refs. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | PTPN11 gen, der koder for det protein tyrosin phosphatase SHP-2., Dette protein er en komponent i adskillige intracellulære signaltransduktionsveje involveret i embryonal udvikling, der modulerer celledeling, differentiering og migration, herunder en medieret af den epidermale vækstfaktorreceptor, som er vigtig i dannelsen af semilunar hjerteklapper. duplikering af kromosomregionen indeholdende PTPN11 kan også resultere i NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
Heterozygote mutationer i de nationale tilsynsmyndigheder, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, og CBL har også været forbundet med en mindre procentdel af NS og relaterede fænotyper.
En tilstand kendt som “neurofibromatosis-Noonan syndrom” er forbundet med neurofibromin.
diagnose
NS kan bekræftes genetisk ved tilstedeværelsen af en af de kendte mutationer, der er anført ovenfor., På trods af identifikation af 14 årsagsgener vil fraværet af en kendt mutation imidlertid ikke udelukke diagnosen, da flere, endnu uopdagede gener kan forårsage NS. Diagnosen af NS er således stadig baseret på kliniske træk. Med andre ord, det er lavet, når en læge føler, at en person har nok af funktionerne til at berettige etiketten. De vigtigste værdier ved at stille en genetisk diagnose er, at den styrer yderligere medicinske og udviklingsmæssige evalueringer, det udelukker andre mulige forklaringer på funktionerne, og det tillader mere nøjagtige estimater af tilbagefaldsrisici., Med flere genotype-fænotype korrelationsundersøgelser, der udføres, vil en positiv genetisk diagnose hjælpe klinikeren med at være opmærksom på mulige anomalier, der er specifikke for den bestemte genmutation. For eksempel ses en stigning i hypertrofisk kardiomyopati hos mennesker med en mutation af KRAS, og der findes en øget risiko for juvenil myelomonocytisk leukæmi for en mutation af ptpn11. I fremtiden kan undersøgelser føre til en målrettet håndtering af NS-symptomer, der afhænger af, hvilken genetisk mutation en person har.,
Før fødslen
Prænatal funktioner, der kan føre læger til at overveje en diagnose af Noonan syndrom omfatter cystisk hygroma, øget nakkestivhed gennemskinnelighed, pleural effusion, og ødem.
differentiel diagnose
mens Turners syndrom har ligheder med nyreanomalier og udviklingsforsinkelse, findes Turners syndrom kun hos kvinder og udtrykker ofte forskelligt. I Turners syndrom er der en lavere forekomst af udviklingsforsinkelser, venstre sidede hjertefejl er konstante, og forekomsten af nyreabnormiteter er meget lavere.,
Andre RASopathies
- Watson syndrom – Watson Syndrom har en række ensartede karakteristika med noonans Syndrom såsom korte statur, pulmonal ventil stenose, variabel intellektuelle udvikling, og hud pigment forandringer.
- Cardiofaciocutan (CFC) syndrom-CFC syndrom ligner meget Noonans syndrom på grund af lignende hjerte-og lymfatiske træk. Men i CFC syndrom intellektuelle handicap og gastrointestinale problemer er ofte mere alvorlige og udtalt.,
- Costello syndrom-lignende CFC syndrom, Costello syndrom har overlappende træk med Noonans syndrom. Imidlertid kan betingelserne skelnes af deres genetiske årsag.
- Neurofibromatosis 1 (NF1)
- Williamilliams syndrom
behandling
behandlingen varierer afhængigt af komplikationer, men har tendens til at være ret standard, hvilket afspejler behandlingen af den generelle befolkning., Ledelsesretningslinjer, divideret med systemer, herunder generel, udviklingsmæssig, dental, vækst og fodring, kardiovaskulær, audiologisk, hæmatologisk, nyre-og skelet, der tegner sig for handlinger, der skal træffes ved diagnose, efter diagnose og hvis symptomatisk, er blevet offentliggjort af et amerikansk konsortium.
specifikt ligner behandling af kardiovaskulære komplikationer den generelle befolkning, og behandling af blødningsdiatese styres af den specifikke faktormangel eller blodpladeaggregering.,
- neuropsykologisk test anbefales for at finde styrker og udfordringer til at skræddersy støtte, der er nødvendig til skole og karriere.
- Uddannelsestilpasning, såsom en individualiseret uddannelsesplan, er undertiden nødvendig for skolebørn.
- taleterapi hvis der er tale-og artikulationsproblemer
- fysioterapi og ergoterapi for brutto – og finmotoriske forsinkelser
- hypotoni og motoriske vanskeligheder påvirker ofte håndskrift. Indkvartering for at mindske håndskrift krav vil forbedre ydeevnen og spare langsigtede håndfunktion.,
- periodisk opfølgning og livslang overvågning af abnormiteter fundet i ethvert system, især det kardiovaskulære system, anbefales.
Anæstesirisiko
selvom nogle få mennesker med Noonan-syndrom er rapporteret at udvikle ondartet hypertermi, er genmutationen af sygdomme, der vides at være forbundet med ondartet hypertermi, anderledes end for Noonan-syndrom.,
Prognose
levetiden for mennesker med noonans syndrom kan være magen til den generelle befolkning, men Noonan syndrom kan være forbundet med flere sundhedsmæssige betingelser, der kan bidrage til dødelighed. Den største bidragyder til dødelighed hos personer med Noonan syndrom er komplikationer af hjerte-kar-sygdom. Prognosen er derfor i høj grad afhængig af tilstedeværelsen eller fraværet af hjertesygdom såvel som sygdommens type og sværhedsgrad (hvis sygdom er til stede)., Mest bemærkelsesværdigt er Noonan syndrom med hypertrofisk kardiomyopati forbundet med øget dødelighed.
Historie
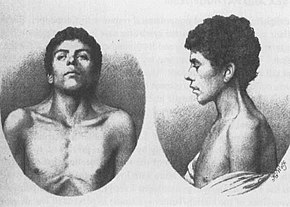
Det ældste kendte tilfælde af NS, beskrevet i 1883 af Kobylinski
Jacqueline Noonan var at øve som en pædiatrisk kardiolog på University of Iowa, da hun bemærkede, at børn med en sjælden form for hjertefejl, valvulær pulmonal stenose, der ofte havde et karakteristisk udseende, med korte statur, svømmehud hals, bredt fordelt øjne, lavt ansatte ører., Både drenge og piger blev ramt. Disse egenskaber blev undertiden set køre i familier, men var ikke forbundet med grove kromosomale abnormiteter. Hun studerede 833 mennesker med Noonan-syndrom på congenital heart disease clinic, på udkig efter andre medfødte abnormiteter, og i 1963 præsenterede hun et papir: “associerede ikke-hjertemisdannelser hos børn med medfødt hjertesygdom”. Dette beskrev 9 børn, der ud over medfødt hjertesygdom havde karakteristiske ansigtstræk, brystdeformiteter og kort statur.Dr. John opit,, tidligere elev af Dr., Noonans, først begyndte at kalde tilstanden “Noonan syndrom”, da han så børn, der lignede dem, som Dr. Noonan havde beskrevet. Dr. Noonan producerede et papir med titlen” Hypertelorism .ith Turner Phenotype ” i 1968, hvor hun studerede 19 patienter, der viste symptomer, der tyder på Noonans syndrom. I 1971 på symposiet om kardiovaskulære defekter blev navnet ‘Noonan syndrom’ officielt anerkendt.
















Skriv et svar