Indledning
Intellektuelle handicap (ID) påvirker næsten 1-2% af befolkningen, og er den mest almindelige neurologiske lidelse (NDD). Et betydeligt antal ID-patienter viser sig at have en genetisk årsag (gennemgået i Bessa et al., 2012)., Genom-dækkende analyse teknikker i øjeblikket anvendes til undersøgelse af ætiologien ofte føre til identifikation af meget sjældne, næsten privat varianter, indsamling af patienter med forandringer i det samme gen, at være et afgørende aspekt af definitionen af et nyt klinisk enhed.
Tidligere i år, patienter, husly mutationer i EBF3 genet er blevet beskrevet, udgør en neuro-udviklingsmæssige syndrom, herunder ID, ataksi, hypotoni, mild ansigts dysmorphisms, og genitourinary abnormiteter (OMIM 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., EBF3-genet (tidlig B-cellefaktor 3) koder for et medlem af den stærkt konserverede tidlige B-cellefaktortranskriptionsfaktorfamilie, udtrykt ved høje niveauer i det udviklende nervesystem (data hentet fra gte. – portalen). EBF3 er et transkriptionelt mål for ar., og vist at være reguleret af NeuroD og ar. (Friocourt og Parnavelas, 2011). AR.koder for en transkriptionsfaktor, der er kritisk for embryonal udvikling, der i mange år har været forbundet med en lang række neuroudviklingsforstyrrelser., Den intellektuelle svækkelse, centralnervesystemet og genitourinære anomalier observeret hos patient med begge mutationer i EBF3 og ar.kan afspejle begge proteiners Bidrag til de samme molekylære og cellulære processer (Chao et al., 2017). EBF3 funktion er også blevet undersøgt i dyremodeller. Ablation af dets ortologer i orme og fluer fører til svækkelse af neuronal udvikling (Prasad et al., 1998; Hattori et al., 2013)., Hos mus fører det at slå Ebf3 ud til neonatal dødelighed og neuronale migrationsdefekter, med svigt i olfaktoriske neuroner projekt til den dorsale olfaktoriske pære (.ang et al., 2004). Den nøjagtige patogene mekanismer EBF3 mutationer er endnu ikke fuldt belyst, men typen af varianter, der er beskrevet hidtil tyder på, at haploinsufficiency, få af funktion, og dominerende negative er muligt, patogene mekanismer for de beskrevne varianter (Chao et al., 2017; Sleven et al., 2017).,
I dette arbejde bidrager vi med en patient med den mindste sletning (600 Kb) rapporteret til dato, der påvirker den samlede EBF3 gen og med en klinisk præsentation overlappende, at patienter med EBF3 single nucleotide varianter (SNVs). Derudover foretager vi en klinisk sammenligning af patienterne med tidligere offentliggjorte store terminale 10q sletninger og rapporterer, at der på trods af forskellene i størrelse er en betydelig fænotypisk overlapning mellem patienter med disse ændringer., Disse fund tilføjer den nuværende viden om EBF3-relaterede lidelser og understøtter EBF3-haploinsufficiens som nøglen i det neuroudviklingssyndrom, der er forbundet med 10terter sletninger.
Materialer og Metoder
patienten blev konstateret i en stor undersøgelse af neurologiske lidelser i Portugal, hvor der er indskrivning af patienter og familier, der blev gjort af den henvisende læge, klinisk information, der var samlet i en anonym database ifølge den portugisiske datatilsynsmyndighed (CNPD) og skriftlig informeret samtykke blev indhentet for alle deltagere., Informeret samtykke til den nuværende patient blev leveret af moderen til den genetiske undersøgelse og offentliggørelse af resultater (inklusive Fotos). Undersøgelsen blev godkendt af det etiske udvalg for Center for Medicinsk Genetik Dr. Jacinto Magalh .es, National Health Institute Dr. Ricardo Jorge.
genomisk DNA blev ekstraheret fra perifert blod under anvendelse af enten Citogen DNA DNA-isolationssæt (Citomed, Portugal). aCGH blev udført ved hjælp af Agilent 180 K array (AMADID:023363) mod en diploid DNA-reference (Kreatech er MegaPoll Reference-DNA, Kreatech Diagnostik, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., Analysen blev gennemført i en 7500-HURTIGT Real-Time PCR-maskine (Applied Biosystems, Foster City, CA, amerikas forenede stater) ved hjælp af Power SYBR Green® (Applied Biosystems, Foster City, CA, amerikas forenede stater) i henhold til producentens anbefalinger og i overensstemmelse med den generelle retningslinjer for qPCR. Specificiteten af hver reaktion blev verificeret ved frembringelsen af en smeltekurve for hvert af de amplificerede fragmenter. Primereffektiviteten blev beregnet ved generering af en standardkurve, der passer til den accepterede normale effektivitetsprocent (anvendte primere anført i supplerende Data)., Ct-værdier opnået for hver test blev analyseret i DataAssist software soft .are (Applied Biosystems, Foster City, CA, USA).
resultater
Her beskriver vi en patient med en de novo-sletning, der påvirker EBF3. Patienten er en 11 år gammel pige med svær ID (global development quuotient = 27 ved 7 år), født af ikke-konsanguineous forældre og uden familiehistorie af neurodevelopmental lidelser. Hun blev født efter en biamniotisk bichorionisk tvillinggraviditet (hendes søster var sund) ved vaginal fødsel ved 35 ugers drægtighed. Fødselsparametre var: vægt, 1.830 g (P3); længde, 42.,5 cm (P10); og OFC, 30,6 cm (P10), med en Apgar score på 8/9 (henholdsvis 1.og 5. min). Den nyfødte periode var kompliceret med sepsis og diagnosen arvelig sfærocytose (arvet fra sin mor). Global udviklingsforsinkelse blev noteret i de første måneder, med hovedkontrol opnået ved 12 måneder, sidder ved 18 måneder, uafhængig gang ved 30 måneder og ingen ord talt i en alder af 3 år., Hun havde pyelonefritis efter 19 måneder (renal ultralyd viste ingen abnormiteter), gastroøsofageal reflu.og tilbagevendende otitis media, med ledende høretab, der krævede kirurgisk indgreb og et høreapparat. Epilepsi blev mistænkt efter 5 måneder (episoder med suspenderet aktivitet), men EEG var normal.
hun blev først observeret i en alder af 3 år 5 måneder (figur 1A,B), på hvilket tidspunkt hun viste muskelhypotoni, hypotonisk ansigt, strabismus og nedsat følsomhed over for smerte., Hun fremlagde også mild dysmorphic funktioner (Figur 1A): trekantede ansigt, lille lav-ører med fremtrædende anti-helix, buede øjenbryn, anteverteret nares, løgformet næse tip, lille mund med downturned hjørner, spidse hage, kort hals, og fremtrædende finger føtal puder, samt en mild kort statur (89 cm, svarende til ca 2SD).

Figur 1. (A) patientens ansigtsudseende efter 3 år og 5 måneder, der viser de små og lavt set ører med fremtrædende anti-Heli.og (B) føtale puder i fingrene., (C) patientens ansigtsudseende ved 11 år. (D) er Fremhævet i grå 600 Kb sletning på 10q26.3-regionen; et zoome ind af EBF3 gen i DGV database afslører eksistensen af 3 sletninger i 3 kontroller, der påvirker de første 6 exons af EBF3 (NM_001005463); CNVs i denne region findes i kontrol befolkninger omfatter udeladelser nvs825626 (til stede i 1/31 personer), nvs552315 (til stede i 1/17421 personer) og nsv552316 (til stede i 1/31 personer). E) den skematiske gengivelse af EBF3-transkripter.,
hjerne MR blev udført efter 6 år, men ingen abnormiteter blev noteret. I en alder af 10 år blev hun revurderet; hun havde stadig tilbagevendende otitis media, men ellers var i god global sundhed. Sprog var meget dårlig (to ord sætninger talt efter 8 år). Hun havde adfærdsmæssige problemer, med stereotype bevægelser (roterende bevægelser, tygge på tøj, hoved retropulsion), hvor han scorede til svær autisme spektrum forstyrrelse (ADI-R og ADOS) på 7 år, hun viste uro og aggressiv adfærd (auto-og hetero) og blev medicineret med antipsykotiske lægemidler., En ortopædisk kirurgi blev udført for pes planus. Ansigtsegenskaberne lignede dem, der tidligere blev beskrevet, med adskilte øvre centrale snit (figur 1C); hun havde briller til strabismus og hypermetropi.
Analyse af genomisk DNA ved aCGH afslørede en de novo-600 kb sletning på 10p26.3 (Figur 1D), der berører tre gener—MGMT (kodning enzymet O-6-methylguanine-DNA methyltransferase, der er involveret i reparation af DNA), EBF3 og GLRX (kodning glutaredoxin, en lille thioltransferase, der fjerner protein GSH addukter), som EBF3 var den mest sandsynlige sygdomme, der er forbundet gen.,
Diskussion
den præsenterede patient blev først analyseret af aCGH for et par år siden. På tidspunktet for aCGH-analyse er eksistensen af de tre varianter, der findes i Database over genomiske varianter (DGV)1, der påvirker de første fem eksoner af EBF3-genet (figur 1E; Park et al., 2010; Cooper et al., 2011), såvel som fraværet af anden kendt sygdom, der forårsager mutationer i dette gen, fører os til at klassificere det som en variant af ukendt betydning (VOUS). Men de seneste publikationer af EBF3 mutationer (Chao et al ., 2017; Harms et al., 2017; Sleven et al.,, 2017) og de kliniske ligheder med de rapporterede tilfælde fører os til at revurdere varianten og få os til at tro, at EBF3-sletning faktisk kan tage højde for sygdommen hos patienten. Et af de aspekter, der er rejst tvivl om den sygdomsfremkaldende af denne variant i første omgang var eksistensen af befolkningen, der styrer bærende sletninger af de første seks exons af dette gen, i heterozygosity (data hentet fra DGV database, som i februar 2017) (Figur 1D). Selvom en transkription af EBF3, der starter i E Exonon13b, er opført i Ensembl-databasen (Enst00000440978.,1) (Figur 1E), som kunne forklare, hvordan sletning af den første exons i sidste ende kunne resultere i en normal fænotype, denne udskrift udelukker DNA-bindende domæne af EBF3, og dens udtryk mønster og funktionelle betydning er ikke blevet karakteriseret. Efter revurdering er de CNV ‘ er, der er beskrevet af Cooper og kolleger (nsv552315 og nsv552316) (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) blev anset for at være på tærsklen til påvisning af SNP microarray og kan ikke danne grundlag for udelukkelse af en kandidat-genet, især i lyset af den stærke genetiske og funktionelle dokumentation for relevansen af EBF3 mutationer til sygdom (Evan Eichler, Greg Bradley Cooper og Coe, personlig kommunikation).,
Vores patient viser, at mange kliniske ligheder med tidligere beskrevne patienter med mutationer i EBF3 (21 sager, der er sammenfattet i Tabel 1), såsom global udviklingsmæssige forsinkelse, forsinket, udtryksfuld tale, hypotoni, øget smerte tærskel, adfærdsmæssige problemer og karakteristiske ansigtstræk (lang/trekantede ansigt, stor pande, hypotonisk ansigt)., Selvom vores patient havde betydelig forsinkelse i motorisk udvikling, blev der imidlertid ikke påvist nogen signifikant ataksi (med nogle begrænsninger i klinisk undersøgelse, da barnet ikke var samarbejdsvilligt), og der var ingen cerebellære anomalier i hjernemri.

Tabel 1. Klinisk sammenligning af det foreliggende tilfælde med de rapporterede tilfælde med punktmutationer/indels i EBF3-genet.
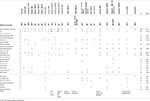
Tabel 2., Klinisk sammenligning af det foreliggende tilfælde med de rapporterede tilfælde med sletninger, der påvirker 102626-cytobåndet.
EBF3 varianter, der er beskrevet i litteraturen i begyndelsen af 2017 omfatter punktmutationer forventes at være skadelige og små insertioner og deletioner, der fører til i ramme sletning af vigtige aminosyrer eller til en frameshift, forudsigeligt forårsager tidlig trunkering af den resulterende protein eller nonsense-medieret decay., De mutationer, der var koncentreret på dele af det gen, der koder for DNA-bindende domæne af EBF3, og blev forudsagt gennem forskellige metoder til at føre til et tab af funktion af denne transskription faktor, hvilket tyder på, nedsat funktion og haploinsufficiency som den mekanisme, der ligger bag neurologiske forstyrrelser hos disse patienter. Knock-out mus til Ebf3 beskrives for at præsentere neonatal dødelighed og neuronale migrationsdefekter, med svigt af olfaktoriske neuroner til at projicere til den dorsale olfaktoriske pære (.ang et al.,, 2004), men ingen beskrivelse er lavet af en fænotype i de Hetero .ygote dyr, som faktisk præsenteres som kontroller i mange af forsøgene, hvilket således ikke understøtter haploinsufficiency-modellen. Vi har gjort en indsats for at opnå og undersøgelse af de neurologiske fænotype af disse dyr, men var ikke vellykket, som den Ebf3(O/E2) knock-out line kan være udgåede (Joseph W. Lewcock, personlig kommunikation). Imidlertid understøtter den aktuelle sag sammen med patienterne opsummeret i tabel 2 hypotesen om EBF3-haploinsufficiens som sygdomsfremkaldende.,
sammenfattende forstærker den aktuelle beskrivelse EBF3-tab af funktion / haploinsufficiens som en årsag til neuro-udviklingssygdom og forstærker sammenhængen mellem dette gen og et karakteristisk klinisk syndrom inden for dette spektrum.
Forfatterbidrag
FL udførte de molekylære undersøgelser og analyserede de molekylære data. MG og GS indsamlede kliniske data. FL, JP og PM gennemgik alle EBF3-mutationssager i litteraturen. FL, GS, JP og PM udarbejdede papiret. PM opnåede finansiering til denne undersøgelse. Undersøgelsen blev udført under ledelse af PM.,
Finansiering
FCT—Fundação para en Ciência e Tecnologia i projekter og stipendier (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Denne artikel er blevet udviklet under rammerne af projektet NORTE-01-0145-FO-000013, der understøttes af den Nordlige Portugal Regionale Operationelle Program (NORTE 2020), under Portugal 2020 partnerskabsaftale, gennem den Europæiske Fond for Regional Udvikling (EFRU).
Erklæring om interessekonflikt
JP var ansat af firmaet CGC Genetics.,
de andre forfattere erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
anerkendelser
Vi vil gerne takke patienten og hendes familie for deltagelse i de genetiske undersøgelser og for at tillade denne publikation. Vi er taknemmelige for, at Dr. Ana Maria Fortuna og Dr. Margarida Reis Lima for at gøre dette projekt muligt, og ved at lette vores samarbejde med CGM.
Fodnoter
1., ^Database over genomiske varianter tilgængelige på: http://dgv.tcag.ca/dgv/app/home .
Friocourt, G., and Parnavelas, J. (2011). Identifikation af AR. – mål afslører nye kandidater til kontrol af kortikal interneuron migration og differentiering. Front. Celle. Neurosci. 5:28. doi: 10.3389/fncel.2011.00028
PubMed Abstract | CrossRef Fuld Tekst | Google Scholar
















Skriv et svar