Masculine sindromul Turner, Noonan-Ehmke sindromul Turner sindromul asemănător, Ullrich-sindromul Noonan
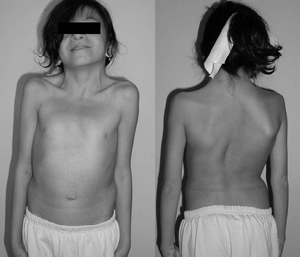
Un copil de 12 ani cu sindromul Noonan. Gât tipic webbed. Curbă structurală dublă cu deformare a coastelor.,irmed cu testarea genetică
Cardiofaciocutaneous sindrom, sindromul Turner, sindromul Costello, neurofibromatoza tip 1
pe Baza simptomelor
hormon de Creștere
Depinde de severitatea problemelor de inimă
1 din 100 (1 în 2000 de boli grave)
sindromul Noonan (NS) este o tulburare genetică care se pot prezenta cu blândețe neobișnuită caracteristici faciale, înălțime scurt, boli cardiace congenitale, probleme cu sangerarea, și malformații scheletice., Caracteristicile faciale includ ochii larg distanțați, ochii de culoare deschisă, urechile joase, gâtul scurt și maxilarul inferior mic. Problemele cardiace pot include stenoza valvei pulmonare. Osul mamar poate să iasă sau să fie scufundat, în timp ce coloana vertebrală poate fi curbată anormal. Inteligența în sindrom este adesea normală. Complicațiile NS pot include leucemie.un număr de mutații genetice pot duce la sindromul Noonan. Condiția poate fi moștenită de la părinții unei persoane ca o condiție dominantă autosomală sau poate apărea ca o nouă mutație., Sindromul Noonan este un tip de Rasopatie, mecanismul de bază pentru care implică supraactivarea în calea de semnalizare a celulelor RAS/MAPK. Diagnosticul poate fi suspectat pe baza simptomelor, a imaginilor medicale și a testelor de sânge. Confirmarea poate fi obținută prin testarea genetică.
nu se cunoaște nici un tratament pentru NS. Tratamentul se bazează pe simptomele și problemele care stau la baza și poate fi necesar un sprijin suplimentar în școală. Terapia cu hormoni de creștere în timpul copilăriei poate crește înălțimea finală a unei persoane afectate. Rezultatele pe termen lung depind de obicei de severitatea problemelor cardiace.,se estimează că 1 din 1000 de persoane sunt ușor afectate de NS, în timp ce aproximativ 1 din 2000 au o formă mai severă a afecțiunii. Masculii par a fi afectați mai des decât femelele. Condiția A fost descrisă pentru prima dată în 1883 și a fost numită după cardiologul pediatric american Jacqueline Noonan, care a descris cazuri suplimentare în 1963.
Semne și simptome

caracteristici Anormale din sindromul Noonan la vârsta de 3 luni: Notă spranceana înclinat și stânga pleoapa scade.,

caracteristici Anormale din sindromul Noonan la vârsta de 3 luni: Notă low-set, posterior rotit, și anomalii de formare ureche. cele mai frecvente semne care conduc la diagnosticarea sindromului Noonan sunt caracteristicile faciale unice și caracteristicile musculo-scheletice. Caracteristicile faciale sunt cele mai proeminente în copilărie, devenind mai puțin evidente odată cu vârsta la multe persoane cu sindrom Noonan.,unele dintre caracteristicile caracteristice ale sindromului Noonan includ un cap mare cu exces de piele pe spatele gâtului, linia părului joasă la nivelul gâtului, linia părului înaltă în partea din față a capului, forma triunghiulară a feței, fruntea largă și un gât scurt, webbed.în ochi, hipertelorismul (ochii larg stabiliți) este o caracteristică definitorie, prezentă la 95% dintre persoanele cu sindrom Noonan., Acest lucru poate fi însoțită de epicanthal falduri (extra pliu de piele la colțul interior al ochiului), ptoza (căderea pleoapelor), proptosis (ochi bulbucați), strabism (activă sau pasivă de cotitură ale ochilor), nistagmus (convulsie mișcarea ochilor) și refracție erori vizuale.
nasul poate fi mic, lat și răsturnat.dezvoltarea gurii poate fi, de asemenea, afectată în sindromul Noonan., Acest lucru poate duce la Filtrum profund canelat (linia buzei superioare) (peste 90%), micrognathia (maxilarul inferior subdimensionat), Palatul arcuit înalt, dificultăți de articulare (dinții nu se aliniază) care pot duce la probleme dentare. Similar cu manifestările musculare de mai sus, în gură, poate fi observat un control slab al limbii.,
Piele
Pielea de semnele și simptomele în sindromul Noonan includ limfedem (limfatici umflarea extremităților), formarea de cheloid, excesiv formarea de cicatrice, hiperkeratoza (supradezvoltare de exterior strat de piele), nevi pigmentari (închise la culoare pete pe piele), și boli ale țesutului conjunctiv
Musculo
Anomalii ale membrelor și extremităților poate apărea în sindromul Noonan. Acest lucru se poate manifesta ca degete cu capăt direct, umplutură suplimentară pe degete și degetele de la picioare, edem al spatelui mâinilor și vârfurilor picioarelor și cubitus valgus (unghi larg de purtare a coatelor).,
pentru statura scurt, hormon de crestere este uneori combinat cu IGF-1 (sau ca o alternativă, IGF-1 ca un stand-alone) poate fi folosit pentru a atinge o înălțime crescută/înălțimea finală mai repede. Înălțimea finală a adulților persoanelor cu sindrom Noonan este de aproximativ 161-167 cm la bărbați și 150-155 cm la femei, ceea ce se apropie de limita inferioară a normalului.anomaliile spinale pot fi prezente până la 30% din timp și acest lucru poate necesita o intervenție chirurgicală pentru a corecta în peste 60% din aceste cazuri., Alte manifestări musculo-scheletice în sindromul Noonan sunt asociate cu tulburări de țesut conjunctiv nediferențiate care pot fi asociate cu contracții articulare (etanșeitate) sau hipermobilitate articulară (slăbiciune). Factori suplimentari pot prezenta sub formă de aripă a scapulei, scolioză, proeminență osoasă a sânului (pectus carinatum), depresie osoasă a sânului (pectus excavatum). Anomaliile musculare se pot prezenta sub formă de hipotonie (tonus muscular scăzut), ceea ce poate duce la lordoză (creșterea cavității în spate) din cauza tonusului muscular abdominal slab.,sindromul Noonan este a doua cea mai frecventă cauză sindromică a bolilor cardiace congenitale. Aceasta include stenoza valvulară pulmonară (50-60%), defecte septale atriale (10-25%), defecte septale ventriculare (5-20%) și cardiomiopatie hipertrofică (12-35%).
plămâni
funcția pulmonară restrictivă a fost raportată la unele persoane.
gastro-intestinale
un număr de diverse simptome gastro-intestinale (GI) au fost asociate cu sindromul Noonan., Acestea includ dificultăți de înghițire, motilitate intestinală scăzută, gastropareză (golire gastrică întârziată), malrotație intestinală și vărsături frecvente sau puternice. Aceste probleme digestive pot duce la scăderea poftei de mâncare, eșecul de a prospera de la copilărie până la pubertate (75%) și, ocazional, necesitatea unui tub de hrănire.{ A fost asociat cu boala Von Willebrand, trombocitopenie Amegacariocitică (număr scăzut de trombocite), timp prelungit de tromboplastină parțială activată, defecte combinate de coagulare., Când sunt prezente, aceste tulburări de însoțire a sindromului Noonan pot fi asociate cu o predispoziție la vânătăi cu ușurință sau hemoragie.uneori, pot apărea malformații Chiari (tip 1), care pot duce la hidrocefalie. De asemenea, au fost raportate convulsii.
Cauze

NS este de obicei moștenit într-un model autosomal dominant cu expresie variabilă.,
recurența la frați și transmiterea aparentă de la părinte la copil a sugerat de mult timp un defect genetic cu moștenire autosomală dominantă și expresie variabilă. Mutațiile în căile de semnalizare a protein kinazei activate Ras/mitogen sunt cunoscute a fi responsabile pentru aproximativ 70% din cazurile de NS.
persoanele cu NS au șanse de până la 50% să le transmită descendenților lor., Faptul că un părinte afectat nu este întotdeauna identificat pentru copii cu NS sugerează mai multe posibilități:
- Manifestari ar putea fi atât de subtilă încât să treacă neobservat (expresivitatea variabilă)
- NS este eterogenă, cuprinzând mai mult de o condiție similară din diferite cauze, iar unele dintre acestea nu pot fi moștenite.
- o proporție mare de cazuri poate reprezenta mutații noi, sporadice.,
| Tip | Online Moștenire Mendeliană la Om bază de date | Gene | An găsit | Locus | % de cazuri | Descriere | Ref. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | A PTPN11 gene codifică protein-tirozin fosfataza SHP-2., Această proteină este o componentă a mai multor căi de transducție a semnalului intracelular implicate în dezvoltarea embrionară care modulează diviziunea, diferențierea și migrarea celulelor, inclusiv una mediată de receptorul factorului de creștere epidermal, care este important în formarea valvelor cardiace semilunare. duplicarea regiunii cromozomiale care conține PTPN11 poate duce, de asemenea, la NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
mutatii Heterozigote în ANR, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, și BIC au fost, de asemenea, asociat cu un procent mai mic de NS și legate de fenotipuri.o afecțiune cunoscută sub numele de” sindrom neurofibromatoză-Noonan ” este asociată cu neurofibromina.
diagnosticul
NS poate fi confirmat genetic prin prezența oricăreia dintre mutațiile cunoscute enumerate mai sus., Cu toate acestea, în ciuda identificării a 14 gene cauzale, absența unei mutații cunoscute nu va exclude diagnosticul, deoarece mai multe gene încă nedescoperite pot provoca NS. Astfel, diagnosticul de NS se bazează încă pe caracteristicile clinice. Cu alte cuvinte, se face atunci când un medic simte că o persoană are suficiente caracteristici pentru a justifica eticheta. Valorile principale ale unui diagnostic genetic sunt că ghidează evaluări medicale și de dezvoltare suplimentare, exclude alte explicații posibile pentru caracteristici și permite estimări mai precise ale riscului de recurență., Cu mai multe studii de corelare genotip-fenotip fiind efectuate, un diagnostic genetic pozitiv va ajuta clinicianul să fie conștient de posibile anomalii specifice acelei anumite mutații genetice. De exemplu, o creștere a cardiomiopatiei hipertrofice este observată la persoanele cu o mutație a KRAS și există un risc crescut de leucemie mielomonocitară juvenilă pentru o mutație a PTPN11. În viitor, studiile pot duce la o gestionare țintită a simptomelor NS care depinde de mutația genetică pe care o are o persoană.,caracteristicile prenatale care ar putea determina medicii să ia în considerare un diagnostic de sindrom Noonan includ higroma chistică, translucența nucală crescută, revărsatul pleural și edemul.în timp ce sindromul Turner are asemănări cu anomaliile renale și întârzierea dezvoltării, sindromul Turner se găsește doar la femei și se exprimă adesea diferit. În sindromul Turner, există o incidență mai mică a întârzierilor de dezvoltare, defectele cardiace la stânga sunt constante, iar apariția anomaliilor renale este mult mai mică.,sindromul Watson – sindromul Watson are o serie de caracteristici similare cu sindromul Noonan, cum ar fi statura scurtă, stenoza valvei pulmonare, dezvoltarea intelectuală variabilă și modificările pigmentului pielii.
Managementul
tratamentul variază în funcție de complicații, dar tind să fie destul de standard, reflectând tratamentul populației generale., Un consorțiu American a publicat ghiduri de Management, împărțite pe sisteme, inclusiv generale, de dezvoltare, dentare, de creștere și de hrănire, cardiovasculare, audiologice, hematologice, renale și scheletice, care iau în considerare acțiunile care trebuie luate la diagnostic, după diagnostic și dacă sunt simptomatice.în mod specific, tratamentul complicațiilor cardiovasculare seamănă cu cel al populației generale, iar tratamentul diatezei hemoragice este ghidat de deficiența factorului specific sau de agregarea plachetară.,testarea neuropsihologică este recomandată pentru a găsi puncte forte și provocări pentru a adapta sprijinul necesar pentru școală și carieră.personalizarea educațională, cum ar fi un plan de program educațional individualizat, este uneori necesară pentru copiii de vârstă școlară.
Istorie
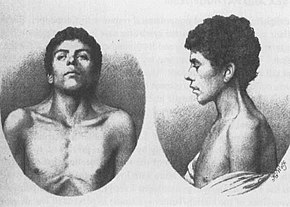
Cel mai vechi caz cunoscut de NS, descrise în 1883 de către Kobyliński
Jacqueline Noonan practica ca un cardiolog pediatru de la Universitatea din Iowa, atunci când ea a observat că copiii cu un tip rar de defect cardiac, valvulară, stenoza pulmonara, de multe ori au un aspect caracteristic fizic, cu statura, palmate gât, larg distanțate ochii, și urechile low-set., Atât băieții, cât și fetele au fost afectate. Aceste caracteristici au fost observate uneori în familii, dar nu au fost asociate cu anomalii cromozomiale brute. A studiat 833 de persoane cu sindrom Noonan la Clinica de boli cardiace congenitale, căutând alte anomalii congenitale, iar în 1963 a prezentat o lucrare: „malformații non-cardiace asociate la copiii cu boli cardiace congenitale”. Acest lucru a descris 9 copii care, în plus față de bolile cardiace congenitale, aveau trăsături caracteristice faciale, deformări toracice și statură scurtă.
Dr. John Opitz, fost student al Dr., Noonan, a început să numească afecțiunea „sindromul Noonan” când a văzut copii care arătau ca cei pe care Dr.Noonan i-a descris. Dr. Noonan a produs o lucrare intitulată „Hypertelorism cu fenotipul Turner” în 1968 unde a studiat 19 pacienți care au prezentat simptome indicative ale sindromului Noonan. În 1971, la Simpozionul de defecte cardiovasculare, numele „sindromul Noonan” a devenit recunoscut oficial.
















Lasă un răspuns