Introducere
dizabilitate Intelectuală (ID) afectează aproximativ 1-2% din populație și este cea mai frecventă afecțiune neurologică (NDD). Se constată că un număr substanțial de pacienți cu ID au o cauză genetică (revizuită în Bessa et al., 2012)., Tehnicile de analiză la nivelul genomului utilizate în prezent pentru investigarea etiologiei conduc adesea la identificarea unor variante aproape private foarte rare, colectarea pacienților cu modificări ale aceleiași gene fiind un aspect crucial al definirii unei noi entități clinice.
la Începutul acestui an, pacienții adăpostirea mutații în EBF3 gene au fost descrise, prezentând un neurodevelopmental sindromul inclusiv ID-ul, ataxie, hipotonie, ușoară faciale dysmorphisms, și genito-urinar anomalii (OMIM 617330) (Chao et al., 2017; Harms și colab., 2017; Sleven și colab., 2017)., La EBF3 (mai Devreme cu celule B Factorul 3) codifică un membru al înalt conservată devreme B-cell factor de transcripție factor de familie, și-a exprimat la un nivel ridicat în țările în curs de sistemul nervos (date preluate din GTEx Portal). EBF3 este o țintă transcripțională a ARX și s-a dovedit a fi reglementată de NeuroD și ARX (Friocourt și Parnavelas, 2011). Arx codifică un factor de transcripție critic pentru dezvoltarea embrionară care, de mulți ani, a fost asociat cu o gamă largă de tulburări de neurodezvoltare., Tulburările intelectuale, anomaliile sistemului nervos central și genitourinar observate la pacienții cu ambele mutații în EBF3 și ARX ar putea reflecta contribuția ambelor proteine la aceleași procese moleculare și celulare (Chao et al., 2017). Funcția FFE 3 a fost, de asemenea, studiată pe modele animale. Ablația ortologilor săi la viermi și muște duce la afectarea dezvoltării neuronale (Prasad et al., 1998; Hattori et al., 2013)., La șoareci, eliminarea Ebf3 duce la letalitate neonatală și defecte de migrație neuronală, cu eșecul proiectului neuronilor olfactivi la bulbul dorsal olfactiv (Wang et al., 2004). Exact mecanismele patogenice de EBF3 mutații nu este încă pe deplin elucidat, dar tipul de variantele descrise până în prezent sugerează că haploinsufficiency, câștig de funcție, și dominant negative sunt posibile mecanisme patogene pentru variantele descrise (Chao et al., 2017; Sleven și colab., 2017).,
În această lucrare vom contribui cu un pacient cu cel mai mic stergere (600 Kb) raportat la data afectează totalitatea EBF3 gene și cu o prezentare clinică se suprapun care a pacienților cu EBF3 single nucleotide variante (SNVs). În plus, facem o comparație clinică a pacienților cu ștergeri mari terminale 10Q publicate anterior și raportăm că, în ciuda diferențelor de dimensiune, există o suprapunere fenotipică semnificativă între pacienții cu aceste modificări., Aceste constatări se adaugă la cunoștințele actuale de EBF3 legate de tulburări și de sprijin EBF3 haploinsufficiency cheie în neurodezvoltare sindrom asociat cu 10qter eliminări.
Materiale și Metode
pacientul A fost constatat într-un studiu de mare de tulburari de neurodezvoltare în Portugalia, în care înscrierea a pacienților și a familiilor a fost făcut de trimitere medic, informații clinice au fost adunate într-un anonim date, conform portugheză Autoritatea pentru Protecția Datelor (CNPD) și în scris consimțământul informat a fost obținut pentru toți participanții., Consimțământul informat pentru pacientul prezent a fost furnizat de mamă pentru studiul genetic și publicarea rezultatelor (inclusiv fotografii). Studiul a fost aprobat de Comitetul de etică al Centrului de Genetică Medicală Dr.Jacinto Magalhães, Institutul Național de sănătate Dr. Ricardo Jorge.
ADN-ul Genomic a fost extras din sângele periferic folosind fie Citogene® kit de izolare ADN (Citomed, Portugalia). aCGH a fost realizată cu ajutorul Agilent 180 K array (AMADID:023363) împotriva unui diploid de ADN de referință (Kreatech e MegaPoll de Referință ADN, Kreatech Diagnosticare, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., Analiza a fost realizată într-o mașină PCR în timp real de 7500 de viteze (Applied Biosystems, Foster City, CA, SUA) utilizând Power SYBR Green® (Applied Biosystems, Foster City, CA, SUA) în conformitate cu recomandările producătorului și respectând liniile directoare generale pentru qPCR. Specificitatea fiecărei reacții a fost verificată prin generarea unei curbe de topire pentru fiecare dintre fragmentele amplificate. Eficiența grundului a fost calculată prin generarea unei curbe standard care să corespundă procentului normal de eficiență acceptat (grundurile utilizate enumerate în datele suplimentare)., Valorile Ct obținute pentru fiecare test au fost analizate în software-ul DataAssist™ (Applied Biosystems, Foster City, CA, USA).
rezultate
aici descriem un pacient cu o deleție de novo care afectează FFE 3. Pacienta este o fetiță de 11 ani cu ID sever (coeficient global de dezvoltare = 27 la vârsta de 7 ani), născută din părinți neconsanguini și fără antecedente familiale de tulburări de neurodezvoltare. S-a născut după o biamniotic bichorionic sarcină gemelară (sora ei a fi sănătos), de livrare vaginale, la 35 de săptămâni de gestație. Parametrii nașterii au fost: greutate, 1830 g (P3); lungime, 42.,5 cm (P10); și OFC, 30,6 cm (P10), cu un scor Apgar de 8/9 (1 și 5 min, respectiv). Perioada neonatală a fost complicată de sepsis și diagnosticul de sferocitoză ereditară (moștenită de la mama ei). Întârzierea dezvoltării globale a fost observată în primele luni, controlul capului fiind realizat la 12 luni, ședința la 18 luni, mersul independent la 30 de luni și fără cuvinte rostite la vârsta de 3 ani., Ea a avut pielonefrită la 19 luni (ecografia renală nu a arătat anomalii), reflux gastroesofagian și otită medie recurentă, cu pierderea auzului conductiv care a necesitat intervenție chirurgicală și un aparat auditiv. Epilepsia a fost suspectată la 5 luni (episoade de activitate suspendată), dar EEG a fost normal.a fost observată pentru prima dată la vârsta de 3 ani și 5 luni (figurile 1A,B), moment în care a prezentat hipotonie musculară, față hipotonică, strabism și sensibilitate redusă la durere., Ea a prezentat, de asemenea, ușoare trăsături dismorfice (Figura 1A): fata triunghiulara, mic low-set cu urechi proeminente anti-helix, sprâncene arcuite, anteverted nares, nas bulbos, gura mică cu downturned colțuri, a subliniat bărbie, gât scurt, și proeminent deget fetale tampoane, precum și o ușoară statura (89 cm, corespunzând la circa 2SD).

Figura 1. (A) aspectul Facial al pacientului la 3 ani și 5 luni care prezintă urechile mici și joase cu anti-helix proeminent și (B) tampoane fetale în degete., (C) aspectul Facial al pacientului la vârsta de 11 ani. (D) Evidențiate în gri 600 Kb stergere la 10q26.3 regiune; un zoom de EBF3 gene în DGV date relevă existența de 3 eliminări în 3 controale care afectează primele 6 exoni de EBF3 (NM_001005463); Cnv în această regiune a fost găsit în controlul populațiilor includ deleții nvs825626 (prezente în 1/31 persoane fizice), nvs552315 (prezente în 1/17421 persoane fizice) și nsv552316 (prezente în 1/31 persoane fizice). (E) Reprezentarea schematică a transcrierilor FFE 3.,RMN-ul cerebral a fost efectuat la 6 ani, dar nu s-au observat anomalii. La vârsta de 10 ani a fost reevaluată; avea încă otită medie recurentă, dar altfel era în stare bună de sănătate globală. Limba a fost foarte slabă (două propoziții de cuvinte vorbite după 8 ani). A avut probleme de comportament, cu mișcări stereotipice (mișcări de rotație, mestecare de haine, retropulsie de cap), scoring pentru tulburare severă de spectru autist (ADI-R și ADOS) la 7 ani; a manifestat agitație și comportament agresiv (auto și hetero) și i s-au administrat medicamente antipsihotice., A fost efectuată o intervenție chirurgicală ortopedică pentru pes planus. Trăsăturile faciale erau similare cu cele descrise anterior, cu incisivi centrali superiori distanțați (figura 1C); avea ochelari pentru strabism și hipermetropie.
Analiza ADN genomic prin aCGH a arătat-o de novo 600 kb stergere la 10p26.3 (Figura 1D) afectează trei gene—MGMT (care codifică O enzimă-6-methylguanine ADN-metiltransferaza, implicate în repararea ADN-ului), EBF3 și GLRX (codare glutaredoxin, un mic thioltransferase care elimină proteine GSH aducți), din care EBF3 a fost cel mai probabil asociată cu boala gene.,
discuție
pacientul prezentat a fost analizat pentru prima dată de aCGH acum câțiva ani. La data de aCGH analiza, existența a trei variante prezente în baza de Date Genomice Variante (DGV)1 afectează primele cinci exonilor EBF3 gene (Figura 1E; Park et al., 2010; Cooper și colab., 2011), precum și absența altor boli cunoscute care cauzează mutații în această genă, ne determină să o clasificăm ca o variantă de semnificație necunoscută (VOUS). Cu toate acestea, publicațiile recente ale mutațiilor EBF3 (Chao et al., 2017; Harms și colab., 2017; Sleven și colab.,, 2017) și asemănările clinice cu cazurile raportate, ne determină să reevaluăm varianta și să ne facă să credem că ștergerea EBF3 poate fi, de fapt, contabilizarea bolii la pacient. Unul dintre aspectele care au ridicat îndoieli cu privire la patogenitatea acestei variante în primul rând a fost existența controalelor populației care poartă ștergeri ale primilor șase exoni ai acestei gene, în heterozigozitate (date preluate din Baza de date DGV din februarie 2017) (figura 1D). Chiar dacă o transcriere a EBF3 începând din Exon13b este listat în baza de date S-a (ENST00000440978.,1) (Figura 1E), ceea ce ar putea explica cum de ștergerea de prima exoni ar putea duce în cele din urmă într-un fenotip normal, această transcriere exclude domeniu de legare a ADN-ului de EBF3, iar expresia sa de model și relevanței funcționale nu au fost caracterizat. La reevaluare, cu toate acestea, Cnv descris de Cooper și colegii (nsv552315 și nsv552316) (Chao et al., 2017; Harms și colab., 2017; Sleven și colab.,, 2017) au fost considerate a fi la pragul de detecție de SNP microarray și nu poate fi baza pentru excluderea unui candidat gene, în special în lumina puternică genetice și funcționale dovezi pentru relevanța EBF3 mutații la boli (Evan Eichler, Greg Cooper și Bradley Coe, comunicare personală).,
pacientul prezinta multe similitudini clinice anterior descrise pacienții cu mutații în EBF3 (21 de cazuri prezentate sintetic în Tabelul 1), cum ar fi global de dezvoltare întârziere, întârziere de vorbire expresiv, hipotonie, creșterea pragului de durere, probleme de comportament și caracteristici faciale caracteristice (long/fata triunghiulara, frunte mare, hipotone fata)., Cu toate acestea, chiar dacă pacientul nostru a avut o întârziere semnificativă în dezvoltarea motorie, nu a fost detectată ataxie semnificativă (cu unele limitări în examenul clinic, deoarece copilul nu a fost cooperant) și nu au fost prezente anomalii cerebeloase în RMN cerebral.

Tabelul 1. Compararea clinică a cazului de față cu cazurile raportate cu mutații punctuale / indels în gena EBF3.
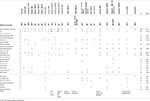
Tabelul 2., Compararea clinică a cazului de față cu cazurile raportate cu ștergeri care afectează cytoband 10q26.
EBF3 variantele descrise în literatura de specialitate la începutul anului 2017 includ mutații punctiforme prezis a fi dăunătoare și mici inserții și ștergeri de conducere în cadrul ștergerea cheie de aminoacizi sau de la un frameshift, previzibil, provocând începutul trunchierea proteinei rezultate sau prostii mediate de degradare., Mutațiile au fost concentrate pe părți din gena care codifică ADN-domeniu de legare a EBF3, și au fost prezise prin diferite metode pentru a duce la o pierdere de funcție de acest factor de transcriere, sugerând astfel funcția redusă și haploinsufficiency ca mecanism de fond al sistemului de tulburări neurodevelopmentale la acești pacienți. Șoarecii Knock-out pentru Ebf3 sunt descriși să prezinte letalitate neonatală și defecte de migrare neuronală, cu eșecul neuronilor olfactivi de a se proiecta la bulbul dorsal olfactiv (Wang et al.,, 2004), dar nu se face nici o descriere a unui fenotip la animalele heterozigote, care sunt de fapt prezentate ca controale în multe dintre experimente, astfel încât să nu susțină modelul haploinsufficiency. Am făcut eforturi pentru a obține și de a studia neurodevelopmental fenotip de aceste animale, dar nu au avut succes, ca Ebf3(O/E2) knock-out linie poate să fi fost întrerupt (Joseph W. Lewcock, comunicare personală). Cu toate acestea, cazul actual, împreună cu pacienții rezumați în tabelul 2, susțin ipoteza HAPLOINSUFFICIENȚEI EBF3 ca boală cauzatoare.,în rezumat, descrierea actuală consolidează pierderea funcției/haploinsufficienței EBF3 ca cauză a bolii neurodevelopmentale și consolidează asocierea acestei gene cu un sindrom clinic caracteristic în acest spectru.
contribuții ale autorului
FL a efectuat studiile moleculare și a analizat datele moleculare. MG și GS au colectat date clinice. FL, JP și PM au revizuit toate cazurile de mutație EBF3 din literatură. FL, GS, JP și PM au elaborat lucrarea. PM a obținut finanțare pentru acest studiu. Studiul a fost realizat sub conducerea PM.,
de Finanțare
FCT—Fundação para un Ciencia e un Tecnologia în proiecte și burse (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Acest articol a fost elaborat în cadrul proiectului NORTE-01-0145-FEDER-000013, susținută de Nord, Portugalia Programul Operațional Regional (NORTE 2020), în Portugalia 2020 Acordul de Parteneriat, prin intermediul Fondului European de Dezvoltare Regională (FEDER).
Declarația privind conflictul de interese
JP a fost angajată de compania CGC Genetics.,ceilalți autori declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.mulțumiri dorim să mulțumim pacientei și familiei sale pentru participarea la studiile genetice și pentru permiterea acestei publicații. Suntem recunoscători pentru Dr. Ana Maria Fortuna și Dr. Margarida Reis Lima pentru a face acest proiect posibil și prin facilitarea colaborării noastre cu CGM.
note de subsol
1., ^Baza de date a variantelor genomice disponibile la: http://dgv.tcag.ca/dgv/app/home.
Friocourt, G., and Parnavelas, J. (2011). Identificarea țintelor Arx dezvăluie noi candidați pentru controlul migrației și diferențierii interneuronului cortical. În față. Celula. Neurosci. 5:28. doi: 10.3389 / fncel.2011.00028
PubMed Abstract / CrossRef Full Text / Google Scholar
















Lasă un răspuns