męski zespół Turnera, zespół Noonana-Ehmke, zespół Turnera-like, zespół Ullricha-Noonana
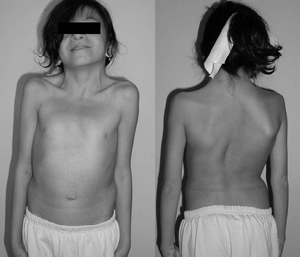
12-letnia dziewczynka z zespołem Noonana. Typowa szyja Pajęcza. Podwójna krzywa strukturalna z deformacją żebra.,irmed z testami genetycznymi
zespół Kardiofaciocutaneous, zespół Turnera, zespół Costello, nerwiakowłókniakowatość typu 1
Na podstawie objawów
hormon wzrostu
zależy od nasilenia problemów z sercem
1 na 100 (1 na 2000 ciężkich chorób)
zespół Noonana (NS) jest zaburzeniem genetycznym, które może objawiać się łagodnymi nietypowymi rysami twarzy, krótkim wzrostem, wrodzoną chorobą serca, problemami z krwawieniami i wadami szkieletu., Rysy twarzy obejmują szeroko rozstawione oczy, jasne oczy, nisko osadzone uszy, krótką szyję i małą dolną szczękę. Problemy z sercem mogą obejmować zwężenie zastawki płucnej. Kość piersi może albo wystają lub zapadnięte, podczas gdy kręgosłup może być nienormalnie zakrzywione. Inteligencja w zespole jest często normalne. Powikłania NS mogą obejmować białaczkę.
wiele mutacji genetycznych może spowodować zespół Noonana. Choroba może być dziedziczona po rodzicach danej osoby jako stan autosomalny dominujący lub wystąpić jako nowa mutacja., Zespół Noonana jest rodzajem Rasopatii, której podstawowym mechanizmem jest nadmierna aktywacja w obrębie szlaku sygnałowego komórek RAS/MAPK. Diagnoza może być podejrzenie na podstawie objawów, obrazowania medycznego, i badania krwi. Potwierdzenie można uzyskać za pomocą badań genetycznych.
nie jest znane lekarstwo na NS. Leczenie opiera się na objawach i Podstawowych Problemów, A Dodatkowe wsparcie w szkole może być wymagane. Terapia hormonem wzrostu w dzieciństwie może zwiększyć dotkniętej osoby ostatecznej wysokości. Długoterminowe wyniki zazwyczaj zależą od ciężkości problemów z sercem.,
szacuje się, że 1 na 1000 osób jest lekko dotkniętych NS, podczas gdy około 1 Na 2000 mA poważniejszą postać choroby. Samce wydają się być dotknięte częściej niż samice. Choroba została po raz pierwszy opisana w 1883 roku i została nazwana na cześć amerykańskiej kardiolog dziecięcej Jacqueline Noonan, która opisała kolejne przypadki w 1963 roku.
objawy przedmiotowe i podmiotowe

nieprawidłowe cechy zespołu Noonana w wieku 3 miesięcy: zwróć uwagę na skośne brwi i opadające powieki po lewej stronie.,

nieprawidłowe cechy zespołu Noonana w wieku 3 miesięcy: zwróć uwagę na nisko osadzone, obrócone tylnie i nieprawidłowo ukształtowane ucho.
najczęstszymi objawami prowadzącymi do rozpoznania zespołu Noonana są unikalne cechy twarzy i cechy układu mięśniowo-szkieletowego. Cechy twarzy są najbardziej widoczne w okresie niemowlęcym, coraz mniej widoczne z wiekiem u wielu osób z zespołem Noonana.,
Głowa
niektóre z charakterystycznych cech zespołu Noonan to duża głowa z nadmiarem skóry z tyłu szyi, niska linia włosów na karku, wysoka linia włosów z przodu głowy, trójkątny kształt twarzy, szerokie czoło i krótka, Pajęcza szyja.
w oczach hiperteloryzm (szeroko osadzone oczy) jest cechą charakterystyczną, występującą u 95% osób z zespołem Noonana., Mogą temu towarzyszyć fałdy epicanthal (dodatkowy fałd skóry w wewnętrznym kąciku oka), opadanie powiek (opadanie powiek), proptosis (wybrzuszone Oczy), zez (odwrócenie do wewnątrz lub na zewnątrz oczu), oczopląs (Szarpanie oczu) i wady wzroku.
nos może być mały, szeroki i odwrócony.
rozwój jamy ustnej może być również zaburzony w zespole Noonana., Może to skutkować głębokim rowkowaniem Filtrum (górna linia wargi) (ponad 90%), mikrognatią (niewymiarowa dolna szczęka), wysokim łukowatym podniebieniem, trudnościami w artykulacji (zęby nie ustawiają się w linii), co może prowadzić do problemów z zębami. Podobnie do powyższych objawów mięśniowych, w jamie ustnej można zaobserwować słabą kontrolę języka.,
Skóra
objawy skóry w zespole Noonan obejmują obrzęk limfatyczny (obrzęk limfatyczny kończyn), powstawanie keloidów, nadmierne powstawanie blizn, hiperkeratoza (nadmierny rozwój zewnętrznej warstwy skóry), znamion barwnikowych (ciemny pigmentowane plamy skóry) i choroby tkanki łącznej
mięśniowo-szkieletowe
nieprawidłowości w kończynach i kończyn mogą wystąpić w zespole Noonan. Może to objawiać się jako bezmyślnie zakończone palce, dodatkowe wyściółki na palcach i palcach, obrzęk z tyłu rąk i czubków stóp, i koślawość łokci (szeroki kąt przenoszenia łokci).,
w przypadku niskiego wzrostu hormon wzrostu jest czasami łączony z IGF-1 (lub jako alternatywa, IGF-1 jako samodzielny) może być stosowany w celu szybszego osiągnięcia zwiększonej wysokości/ostatecznej wysokości. Ostateczny wzrost dorosłego osobnika z zespołem Noonana wynosi około 161-167 cm u mężczyzn i 150-155 cm u kobiet, co zbliża się do dolnej granicy normy.
nieprawidłowości kręgosłupa mogą występować do 30% czasu i może to wymagać operacji w celu skorygowania w ponad 60% tych przypadków., Inne objawy mięśniowo-szkieletowe w zespole Noonana są związane z niezróżnicowanymi zaburzeniami tkanki łącznej, które mogą być związane z przykurczami stawów (ucisk) lub nadmierną ruchliwością stawów (luźność). Dodatkowe czynniki mogą występować w postaci zwichnięcia łopatki, skoliozy, wyeksponowania kości piersi( pectus carinatum), depresji kości piersi (Pectus excavatum). Zaburzenia mięśniowe mogą występować jako hipotonia (niskie napięcie mięśniowe), co może prowadzić do lordozy (zwiększone zagłębienie w plecach) z powodu słabego napięcia mięśni brzucha.,
Serce
zespół Noonana jest drugą najczęstszą przyczyną wrodzonej choroby serca. Obejmuje to zwężenie zastawki płucnej (50-60%), ubytki przegrody przedsionkowej (10-25%), ubytki przegrody komorowej (5-20%) i kardiomiopatię przerostową (12-35%).
płuca
żołądkowo-jelitowe
z zespołem Noonana wiąże się wiele różnorodnych objawów żołądkowo-jelitowych (GI)., Należą do nich trudności w połykaniu, mała ruchliwość jelit, gastropareza( opóźnione opróżnianie żołądka), malrotacja jelit i częste lub silne wymioty. Te problemy trawienne mogą prowadzić do zmniejszenia apetytu, niepowodzenie rozwijać się od niemowlęctwa do dojrzewania (75%), a czasami potrzeba rurki do karmienia.{ Jest związana z chorobą Von Willebranda, małopłytkowością Amegakaryocytarną (niską liczbą płytek krwi), wydłużonym czasem częściowej tromboplastyny po aktywacji, złożonymi wadami krzepnięcia., Gdy obecne, te Noonan zespół towarzyszące zaburzenia mogą być związane z predyspozycją do siniaków łatwo, lub krwotoku.
neurologiczne
sporadycznie może wystąpić malformacja Chiariego (typ 1), która może prowadzić do wodogłowia. Zgłaszano również napady padaczkowe.
powoduje

NS jest zazwyczaj dziedziczone w autosomalnym dominującym wzorze z wyrażeniem zmiennej.,
nawrót u rodzeństwa i pozorna transmisja z rodzica na dziecko od dawna sugerują defekt genetyczny z dziedziczeniem autosomalnym dominującym i zmienną ekspresją. Mutacje w szlakach sygnałowych kinazy białkowej aktywowanej Ras/mitogenem są odpowiedzialne za około 70% przypadków NS.
osoby z NS mają do 50% szans na przekazanie go potomstwu., Fakt, że dotknięty rodzic nie zawsze jest identyfikowany dla dzieci z NS sugeruje kilka możliwości:
- manifestacje mogą być tak subtelne, że nie są rozpoznawane (zmienna ekspresja)
- NS jest niejednorodna, zawierająca więcej niż jeden podobny stan o różnych przyczynach, a niektóre z nich mogą nie być dziedziczone.
- duża część przypadków może reprezentować nowe, sporadyczne mutacje.,
| Typ | dziedziczenie Mendla online w Bazie Danych człowieka | Gen | rok znaleziony | Locus | % przypadków | opis | ref. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | Gen PTPN11 koduje białkową fosfatazę tyrozynową SHP-2., Białko to jest składnikiem kilku wewnątrzkomórkowych szlaków transdukcji sygnału zaangażowanych w rozwój embrionalny, które modulują podział komórek, różnicowanie i migrację, w tym jeden pośredniczy w receptor naskórkowego czynnika wzrostu, który jest ważny w tworzeniu semilunar zastawek serca. duplikacja regionu chromosomu zawierającego PTPN11 może również prowadzić do NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
heterozygotyczne mutacje w NRAS, HRAS, BRAF, shoc2, map2k1, map2k2 i CBL również były związane z mniejszym odsetkiem NS i pokrewnych fenotypów.
stan znany jako „nerwiakowłókniakowatość-zespół Noonana” jest związany z nerwiakowłókniakowatością.
rozpoznanie
NS może być potwierdzone genetycznie przez obecność którejkolwiek ze znanych mutacji wymienionych powyżej., Jednak pomimo identyfikacji 14 genów przyczynowych, brak znanej mutacji nie wyklucza diagnozy, ponieważ więcej, jak dotąd-nieodkrytych genów może powodować NS. Tak więc diagnoza NS nadal opiera się na cechach klinicznych. Innymi słowy, robi się to, gdy lekarz uważa, że dana osoba ma wystarczająco dużo cech, aby zagwarantować Etykietę. Główne wartości dokonywania diagnozy genetycznej są to, że prowadzi dodatkowe oceny medyczne i rozwojowe, wyklucza inne możliwe wyjaśnienia dla cech, i pozwala bardziej dokładne szacunki ryzyka nawrotu., Przy przeprowadzaniu większej liczby badań korelacji genotyp-fenotyp, pozytywna diagnoza genetyczna pomoże lekarzowi zdawać sobie sprawę z możliwych anomalii specyficznych dla danej mutacji genu. Na przykład wzrost kardiomiopatii przerostowej obserwuje się u osób z mutacją KRAS i istnieje zwiększone ryzyko młodzieńczej białaczki mielomonocytowej dla mutacji PTPN11. W przyszłości badania mogą prowadzić do ukierunkowanego zarządzania objawami NS, które zależą od tego, jaką mutację genetyczną ma dana osoba.,
przed porodem
cechy prenatalne, które mogą skłonić lekarzy do rozważenia diagnozy zespołu Noonana, obejmują torbielowatą higromę, zwiększoną przezierność karku, wysięk opłucnowy i obrzęk.
diagnostyka różnicowa
podczas gdy zespół Turnera ma podobieństwa z anomaliami nerek i opóźnieniem rozwoju, zespół Turnera występuje tylko u kobiet i często wyraża się inaczej. W zespole Turnera występuje mniejsza częstość opóźnień rozwojowych, lewostronne wady serca są stałe, a występowanie zaburzeń czynności nerek jest znacznie mniejsze.,
Inne Rasopatie
- zespół Watsona – zespół Watsona ma wiele podobnych cech z zespołem Noonana, takich jak niski wzrost, zwężenie zastawki płucnej, zmienny rozwój intelektualny i zmiany barwnikowe skóry.
- zespół Cardiofaciocutaneous (CFC) – zespół CFC jest bardzo podobny do zespołu Noonana ze względu na podobne cechy sercowe i limfatyczne. Jednak w zespole CFC niepełnosprawność intelektualna i problemy żołądkowo-jelitowe są często bardziej poważne i wyraźne.,
- zespół Costello-podobnie jak zespół CFC, zespół Costello ma cechy pokrywające się z zespołem Noonana. Jednak warunki można odróżnić od ich przyczyny genetycznej.
- nerwiakowłókniakowatość 1 (NF1)
- zespół Williamsa
Zarządzanie
leczenie różni się w zależności od powikłań, ale wydają się być dość standardowe, odzwierciedlając leczenie populacji ogólnej., Wytyczne zarządzania, podzielone na systemy, w tym ogólne, rozwojowe, stomatologiczne, wzrost i karmienie, sercowo-naczyniowe, audiologiczne, hematologiczne, nerkowe i szkieletowe, które uwzględniają działania, które należy podjąć przy diagnozie, po diagnozie i jeśli są objawowe, zostały opublikowane przez amerykańskie konsorcjum.
w szczególności leczenie powikłań sercowo-naczyniowych przypomina leczenie populacji ogólnej, a leczenie skazy krwotocznej opiera się na specyficznym niedoborze czynnika lub agregacji płytek krwi.,
- dostosowanie edukacyjne, takie jak zindywidualizowany plan programu edukacyjnego, jest czasami potrzebne dzieciom w wieku szkolnym.
- terapia logopedyczna jeśli występują problemy z wymową i artykulacją
- fizykoterapia i terapia zajęciowa w przypadku opóźnień ruchowych i drobnych
- hipotonia i trudności ruchowe często wpływają na charakter pisma. Przystosowanie do zmniejszania wymagań pisma ręcznego poprawi wydajność i pozwoli zaoszczędzić długoterminową funkcję dłoni.,
- zaleca się okresową obserwację i dożywotnie monitorowanie nieprawidłowości występujących w każdym układzie, szczególnie w układzie sercowo-naczyniowym.
ryzyko znieczulenia
chociaż u kilku osób z zespołem Noonana stwierdzono wystąpienie hipertermii złośliwej, mutacja genowa chorób, o których wiadomo, że są związane z hipertermią złośliwą, różni się od zespołu Noonana.,
rokowanie
długość życia osób z zespołem Noonana może być podobna do populacji ogólnej, jednak zespół Noonana może być związany z kilkoma schorzeniami, które mogą przyczynić się do śmiertelności. Największym czynnikiem śmiertelności u osób z zespołem Noonana są powikłania chorób układu krążenia. Rokowanie jest więc w dużej mierze zależne od obecności lub braku choroby serca, a także rodzaju i ciężkości choroby(jeśli choroba jest obecna)., Przede wszystkim zespół Noonana z kardiomiopatią przerostową wiąże się ze zwiększoną śmiertelnością.
Historia
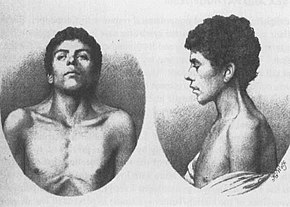
Jacqueline Noonan praktykowała jako kardiolog dziecięcy w University of Iowa, kiedy zauważyła, że dzieci z rzadkim typem wady serca, zwężenie zastawki płucnej, często miały charakterystyczny wygląd fizyczny, z niskim wzrostem, szyjką w pasie, szeroko rozstawionymi oczami i nisko osadzonymi uszami., Dotyczyło to zarówno chłopców, jak i dziewcząt. Cechy te były czasami obserwowane w rodzinach, ale nie były związane z poważnymi nieprawidłowościami chromosomalnymi. Badała 833 osoby z zespołem Noonana w klinice wad wrodzonych serca, szukając innych wad wrodzonych, a w 1963 przedstawiła pracę: „Associated non-cardiac deformations in children with congenital heart disease”. Opisano 9 dzieci, które oprócz wrodzonej choroby serca miały charakterystyczne rysy twarzy, deformacje klatki piersiowej i niski wzrost.
Dr John Opitz, były student Dr, Noonan po raz pierwszy zaczął nazywać stan „syndromem Noonana”, gdy zobaczył dzieci, które wyglądały jak te, które opisał Dr Noonan. Dr Noonan wyprodukowała pracę zatytułowaną „Hyperteloryzm z fenotypem Turnera” w 1968 roku, w której zbadała 19 pacjentów, którzy wykazywali objawy wskazujące na zespół Noonana. W 1971 roku na Sympozjum chorób układu krążenia oficjalnie uznano nazwę „zespół Noonana”.
















© 2021 Tombouctou
Theme by Anders Noren — Up ↑
Dodaj komentarz