wprowadzenie
niepełnosprawność intelektualna (ID) dotyka prawie 1-2% populacji i jest najczęstszym zaburzeniem neurorozwojowym (NDD). Znaczna liczba pacjentów ID stwierdzono, że mają przyczynę genetyczną(reviewed in Bessa et al., 2012)., Techniki analizy genomu stosowane obecnie do badania etiologii często prowadzą do identyfikacji bardzo rzadkich, niemal prywatnych wariantów, przy czym zbiór pacjentów ze zmianami w tym samym Genie jest kluczowym aspektem definicji nowego podmiotu klinicznego.
na początku tego roku opisano pacjentów z mutacjami w genie EBF3, prezentujących zespół neurorozwojowy, w tym ID, ataksja, hipotonia, łagodne dysmorfizmy twarzy i nieprawidłowości układu moczowo-płciowego (OMIM 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., Gen EBF3 (Early B cell Factor 3) koduje członka wysoko zachowanej wczesnej rodziny czynników transkrypcyjnych czynnika B, wyrażonej na wysokim poziomie w rozwijającym się układzie nerwowym (dane pobrane z portalu GTEx). EBF3 jest transcriptionalnym celem ARX i wykazano, że jest regulowany przez Neuroda i ARX (Friocourt and Parnavelas, 2011). ARX koduje czynnik transkrypcyjny krytyczny dla rozwoju zarodkowego, który przez wiele lat był związany z szerokim zakresem zaburzeń neurorozwojowych., Upośledzenie intelektualne, ośrodkowy układ nerwowy i anomalie układu moczowo-płciowego obserwowane u pacjenta z obu mutacji w EBF3 i ARX może odzwierciedlać wkład obu białek do tych samych procesów molekularnych i komórkowych (Chao et al., 2017). Funkcję EBF3 badano również w modelach zwierzęcych. Ablacja jego ortologów w robakach i muchach prowadzi do upośledzenia rozwoju neuronalnego (Prasad et al., 1998; Hattori et al., 2013)., U myszy, wybijanie Ebf3 prowadzi do śmiertelności noworodków i neuronalnych wad migracji, z niepowodzeniem projektu neuronów węchowych do grzbietowej żarówki węchowej (Wang et al., 2004). Dokładne patogenne mechanizmy mutacji EBF3 nie jest jeszcze w pełni wyjaśnione, ale Typ wariantów opisanych do tej pory sugerują, że haploinsufficiency, gain of function i dominujący negatywny są możliwymi patogennymi mechanizmami dla opisanych wariantów (Chao et al., 2017; Sleven et al., 2017).,
w pracy tej uczestniczymy z pacjentem z najmniejszą delecją (600 Kb) zgłoszoną do tej pory wpływającą na całość genu EBF3 oraz z prezentacją kliniczną pokrywającą się z prezentacją pacjentów z pojedynczymi wariantami nukleotydów EBF3 (SNVs). Ponadto dokonujemy porównania klinicznego pacjentów z wcześniej opublikowanymi dużymi delecjami terminala 10q i informujemy, że pomimo różnic w wielkości, istnieje znaczne nakładanie się fenotypów między pacjentami z tymi zmianami., Wyniki te uzupełniają aktualną wiedzę na temat zaburzeń związanych z EBF3 i wspierają niedobór haploinsufficiency EBF3 jako kluczowy w zespole neurorozwojowym związanym z delecjami 10qter.
materiały i metody
pacjent został ustalony w ramach dużego badania zaburzeń neurorozwojowych w Portugalii, w którym rejestracja pacjentów i rodzin została dokonana przez lekarza kierującego, informacje kliniczne zostały zebrane w anonimowej bazie danych zgodnie z portugalskim Urzędem Ochrony Danych (CNPD) i uzyskano pisemną świadomą zgodę dla wszystkich uczestników., Świadoma zgoda dla obecnego pacjenta została dostarczona przez matkę do badania genetycznego i publikacji wyników (w tym zdjęć). Badanie zostało zatwierdzone przez komitet etyki Centrum Genetyki Medycznej Dr Jacinto Magalhães, Narodowy Instytut Zdrowia dr Ricardo Jorge.
genomowe DNA zostało wyekstrahowane z krwi obwodowej za pomocą zestawu do izolacji DNA Citogene® (Citomed, Portugalia). aCGH wykonano przy użyciu tablicy Agilent 180 K (AMADID:023363) przeciwko diploidalnemu DNA referencyjnemu (Kreatech ' s MegaPoll Reference DNA, Kreatech Diagnostics, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., Analizę przeprowadzono na maszynie PCR o szybkości 7500 W Czasie Rzeczywistym (Applied Biosystems, Foster City, CA, USA) z wykorzystaniem Power SYBR Green® (Applied Biosystems, Foster City, CA, USA), zgodnie z zaleceniami producenta i ogólnymi wytycznymi dla qPCR. Specyficzność każdej reakcji została zweryfikowana poprzez wygenerowanie krzywej topnienia dla każdego ze wzmocnionych fragmentów. Sprawność gruntowania została obliczona poprzez wygenerowanie krzywej standardowej odpowiadającej przyjętemu procentowi sprawności normalnej (stosowane grunty wymienione w danych uzupełniających)., Wartości Ct uzyskane dla każdego testu zostały przeanalizowane w oprogramowaniu DataAssist™ (Applied Biosystems, Foster City, CA, USA).
wyniki
tutaj opisujemy pacjenta z De novo delecją wpływającą na EBF3. Pacjentem jest 11-letnia dziewczynka z ciężkim ID (global development quotient = 27 at 7 years of age), Urodzona z niedojrzałych rodziców i bez wywiadu rodzinnego zaburzeń neurorozwojowych. Urodziła się po dwuliściennej ciąży bliźniaczej (jej siostra jest zdrowa), przez poród dopochwowy, w 35 tygodniu ciąży. Parametry urodzeniowe to: waga 1830 g (P3); Długość 42.,5 cm (P10); oraz OFC, 30,6 cm (P10), z wynikiem Apgar 8/9 (odpowiednio 1.i 5. min). Okres noworodkowy był skomplikowany z posocznicą i rozpoznaniem dziedzicznej sferocytozy (odziedziczonej po matce). W pierwszych miesiącach odnotowano globalne opóźnienie rozwojowe, z kontrolą głowy osiąganą po 12 miesiącach, siedzeniem po 18 miesiącach, samodzielnym chodzeniem po 30 miesiącach i brakiem słów w wieku 3 lat., Miała odmiedniczkowe zapalenie nerek w wieku 19 miesięcy (USG nerek nie wykazało nieprawidłowości), refluks żołądkowo-przełykowy i nawracające zapalenie ucha środkowego, z przewodzeniowym ubytkiem słuchu, które wymagało interwencji chirurgicznej i aparatu słuchowego. Epilepsję podejrzewano po 5 miesiącach (epizody zawieszonej aktywności), ale EEG było w normie.
Po raz pierwszy zaobserwowano ją w wieku 3 lat 5 miesięcy (ryc. 1A,B), w tym czasie wykazywała hipotonię mięśniową, hipotoniczną twarz, zeza i zmniejszoną wrażliwość na ból., 1A): trójkątna twarz, Małe nisko osadzone uszy z widoczną anty helisą, łukowate brwi, przedplecze, bulwiasty czubek nosa, małe usta z odwróconymi kącikami, spiczasty podbródek, krótka szyja i wydatne podkładki pod palce płodu, a także łagodny niski wzrost (89 cm, co odpowiada około 2SD).

Rysunek 1. A) wygląd twarzy pacjenta w wieku 3 lat i 5 miesięcy, pokazujący małe i nisko osadzone uszy z widocznymi przeciwpierścieniami oraz b) podkładki płodowe w palcach., C) wygląd twarzy pacjenta w wieku 11 lat. (D) zaznaczona na szaro delecja 600 Kb w regionie 10q26.3; powiększenie genu EBF3 w bazie danych DGV ujawnia istnienie 3 delecji w 3 kontrolach, które wpływają na pierwsze 6 eksonów EBF3 (NM_001005463); CNV w tym regionie stwierdzone w populacjach kontrolnych obejmują delecje nvs825626 (obecne u 1/31 osobników), nvs552315 (obecne u 1/17421 osobników) i NVS552315 (obecne u 1/17421 osobników).nsv552316 (występuje u 1/31 osobników). E) schematyczne przedstawienie transkryptów EBF3.,
rezonans mózgu wykonano w wieku 6 lat, ale nie stwierdzono żadnych nieprawidłowości. W wieku 10 lat została ponownie oceniona; nadal miała nawracające zapalenie ucha środkowego, ale poza tym była w dobrym stanie światowym. Język był bardzo ubogi(dwa zdania wypowiedziane po 8 latach). Miała problemy z zachowaniem, ze stereotypowymi ruchami (ruchy obrotowe, żucie ubrań, retropulsja głowy), punktacją na ciężkie zaburzenia ze spektrum autyzmu (ADI-R i ADOS) w wieku 7 lat; wykazywała pobudzenie i agresywne zachowanie (auto i hetero) i była leczona lekami przeciwpsychotycznymi., Przeprowadzono operację ortopedyczną na pes planus. Rysy twarzy były podobne do tych wcześniej opisanych, z rozstawionymi górnymi centralnymi siekaczami( ryc. 1C); miała okulary dla zeza i hipermetropii.
Analiza genomowego DNA przez aCGH wykazała delecję de novo 600 kb przy 10p26 .3 (Rysunek 1D) wpływającą na trzy geny-MGMT (kodujący enzym o-6-metyloguanino-DNA metylotransferazy, biorący udział w naprawie DNA), EBF3 i GLRX (kodujący glutaredoksynę, małą tioltransferazę, która usuwa addukty białka GSH), z których EBF3 był najbardziej prawdopodobnym genem związanym z chorobą.,
dyskusja
prezentowany pacjent został po raz pierwszy przeanalizowany przez aCGH kilka lat temu. W czasie analizy aCGH, istnienie trzech wariantów obecnych w Database of genomic Variants (DGV) 1 wpływających na pierwsze pięć eksonów genu EBF3 (rysunek 1e; Park et al., 2010; Cooper et al., 2011), a także brak innych znanych chorób powodujących mutacje w tym genie, skłania do zaklasyfikowania go jako wariantu o nieznanym znaczeniu (VOUS). Jednak ostatnie publikacje mutacji EBF3 (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) i kliniczne podobieństwa ze zgłaszanymi przypadkami, prowadzą nas do ponownej oceny wariantu i sprawiają, że uważamy, że delecja EBF3 może w rzeczywistości uwzględniać chorobę u pacjenta. Jednym z aspektów, który budził wątpliwości co do patogenności tego wariantu w pierwszej kolejności, było istnienie populacji kontrolującej delecje pierwszych sześciu eksonów tego genu w heterozygotyczności (dane pobrane z bazy danych DGV z lutego 2017 r.) (ryc. 1D). Mimo że zapis EBF3 zaczynający się w Exon13b jest wymieniony w bazie danych Ensembl (ENST00000440978.,1) (Rysunek 1E), które mogłyby wyjaśnić, w jaki sposób usunięcie pierwszych eksonów może ostatecznie doprowadzić do normalnego fenotypu, transkrypt ten wyklucza domenę wiążącą DNA EBF3, a jego wzór ekspresji i znaczenie funkcjonalne nie zostały scharakteryzowane. Po ponownej ocenie, jednak CNVs opisane przez Cooper i współpracowników (nsv552315 i nsv552316) (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) zostały uznane za znajdujące się na progu wykrycia przez SNP microarray i nie mogą być podstawą do wykluczenia genu kandydującego, szczególnie w świetle silnych dowodów genetycznych i funkcjonalnych na znaczenie mutacji EBF3 dla choroby (Evan Eichler, Greg Cooper i Bradley Coe, personal communication).,
nasz pacjent wykazuje wiele podobieństw klinicznych z wcześniej opisanymi pacjentami z mutacjami w EBF3 (21 przypadków podsumowanych w tabeli 1), takimi jak globalne opóźnienie rozwojowe, opóźniona mowa ekspresyjna, hipotonia, podwyższony próg bólu, problemy behawioralne i charakterystyczne rysy twarzy (długa / trójkątna twarz, duże czoło, hipotoniczna twarz)., Jednak mimo znacznego opóźnienia w rozwoju ruchowym, nie wykryto istotnej ataksji (z pewnymi ograniczeniami w badaniu klinicznym, gdyż dziecko nie współpracowało), a w MRI mózgu nie stwierdzono nieprawidłowości móżdżku.

Tabela 1. Porównanie kliniczne obecnego przypadku z opisanymi przypadkami z mutacjami punktowymi / indelami w genie EBF3.
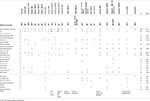
Tabela 2., Porównanie kliniczne obecnego przypadku ze zgłoszonymi przypadkami delecji wpływającymi na cytoband 10q26.
warianty EBF3 opisane w literaturze na początku 2017 roku obejmują mutacje punktowe przewidywane jako szkodliwe i małe insercje i delecje prowadzące do delecji kluczowych aminokwasów w ramce lub do zmiany ramki, co przewidywalnie powoduje wczesne obcięcie powstałego białka lub rozpad nonsensowny., Mutacje koncentrowały się na częściach genu kodującego domenę wiążącą DNA EBF3 i przewidywano za pomocą różnych metod, że doprowadzą do utraty funkcji tego czynnika transkrypcyjnego, sugerując tym samym zmniejszoną funkcję i niedobór haploinsufficiency jako mechanizm leżący u podstaw zaburzeń neurorozwojowych u tych pacjentów. Knock-out myszy dla Ebf3 są opisane, aby przedstawić noworodków śmiertelność i neuronów migracji defekty, z niepowodzeniem neuronów węchowych do projektu do grzbietowej żarówki węchowej (Wang et al.,, 2004), ale nie ma opisu fenotypu u zwierząt heterozygotycznych, które są faktycznie prezentowane jako kontrolki w wielu eksperymentach, nie wspierając tym samym modelu haploinsufficiency. Podjęliśmy wysiłki w celu uzyskania i zbadania fenotypu neurorozwojowego tych zwierząt, ale nie powiodło się, ponieważ linia nokautu Ebf3(o/E2) mogła zostać przerwana (Joseph W. Lewcock, personal communication). Jednak obecny przypadek wraz z pacjentami podsumowanymi w tabeli 2, popierają hipotezę haploinsufficiency EBF3 jako choroby powodującej.,
podsumowując, obecny opis wzmacnia utratę funkcji/haploinsufficiency EBF3 jako przyczynę choroby neurorozwojowej i wzmacnia skojarzenie tego genu z charakterystycznym zespołem klinicznym w tym spektrum.
wkład autora
MG i GS zebrały dane kliniczne. FL, JP i PM przejrzały wszystkie przypadki mutacji EBF3 w literaturze. FL, GS, JP i PM przygotowały gazetę. PM uzyskał dofinansowanie na to badanie. Badanie przeprowadzono pod kierunkiem PM.,
finansowanie
w ramach projektów i stypendiów (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Artykuł został opracowany w ramach projektu NORTE-01-0145-FEDER-000013, wspierany przez Regionalny Program Operacyjny północnej Portugalii (NORTE 2020), w ramach umowy o partnerstwie Portugalia 2020, za pośrednictwem Europejskiego Funduszu Rozwoju Regionalnego (FEDER).
,
pozostali autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
podziękowania
dziękujemy pacjentce i jej rodzinie za udział w badaniach genetycznych oraz za umożliwienie publikacji. Jesteśmy wdzięczni Dr Ana Maria Fortuna i dr Margarida Reis Lima za umożliwienie realizacji tego projektu i ułatwienie naszej współpracy z CGM.
Przypisy
1., W 1997 roku w Polsce wprowadzono do użytku nową linię produkcyjną.
Friocourt, G., and Parnavelas, J. (2011). Identyfikacja celów Arx ujawnia nowych kandydatów do kontrolowania migracji i różnicowania interneuronów korowych. Przód. Cell. Neurosci. 5:28. podoba mi się! do obserwowanych nr: 1033892011.00028
PubMed Abstract | CrossRef Full Text | Google Scholar
















Dodaj komentarz