Introduction
intellectuele handicap (ID) treft bijna 1-2% van de bevolking en is de meest voorkomende neurodevelopmental disorder (NDD). Een substantieel aantal ID-patiënten worden gevonden om een genetische oorzaak te hebben (beoordeeld in Bessa et al., 2012)., Genoombrede analysetechnieken die momenteel worden gebruikt voor onderzoek van de etiologie leiden vaak tot de identificatie van zeer zeldzame bijna private varianten, waarbij het verzamelen van patiënten met veranderingen in hetzelfde gen een cruciaal aspect is van de definitie van een nieuwe klinische entiteit.
eerder dit jaar werden patiënten met mutaties in het EBF3-gen beschreven, met een neurologisch ontwikkelingssyndroom waaronder ID, ataxie, hypotonie, lichte faciale dysmorfismen en urogenitale afwijkingen (Omim 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., Het EBF3-gen (Early B celfactor 3) codeert voor een lid van de sterk geconserveerde early B-celfactortranscriptiefactorfamilie, die op hoge niveaus in het zich ontwikkelende zenuwstelsel wordt uitgedrukt (gegevens die van Gtex-portaal worden opgehaald). EBF3 is een transcriptioneel doel van ARX, en aangetoond te worden gereguleerd door NeuroD en ARX (Friocourt en Parnavelas, 2011). ARX codeert een transcriptiefactor die cruciaal is voor embryonale ontwikkeling en die al vele jaren in verband wordt gebracht met een breed scala aan neurologische ontwikkelingsstoornissen., De intellectuele stoornis, het centrale zenuwstelsel en urogenitale anomalieën waargenomen bij patiënten met beide mutaties in EBF3 en ARX kunnen de bijdrage van beide eiwitten aan dezelfde moleculaire en cellulaire processen weerspiegelen (Chao et al., 2017). De functie EBF3 is ook bestudeerd in dierlijke modellen. Ablatie van zijn orthologs in wormen en vliegen leidt tot verslechtering van neuronale ontwikkeling (Prasad et al., 1998; Hattori et al., 2013)., Bij muizen leidt het uitschakelen van Ebf3 tot neonatale letaliteit en neuronale migratiedefecten, met falen van olfactorische neuronen project naar de dorsale olfactorische bol (Wang et al., 2004). De exacte pathogene mechanismen van EBF3 mutaties zijn nog niet volledig opgehelderd, maar het type van varianten tot nu toe beschreven suggereren dat haploinsufficižntie, winst van functie, en dominant negatief zijn mogelijke pathogene mechanismen voor de beschreven varianten (Chao et al., 2017; Sleven et al., 2017).,
in dit werk dragen we bij met een patiënt met de kleinste deletie (600 Kb) die tot op heden is gemeld en die de totaliteit van het ebf3-gen beïnvloedt en met een klinische presentatie die die van patiënten met EBF3 enkelvoudige nucleotidevarianten (SNVs) overlapt. Daarnaast maken we een klinische vergelijking van de patiënten met eerder gepubliceerde grote terminale 10Q-deleties en melden we dat er, ondanks de verschillen in grootte, een significante fenotypische overlap is tussen patiënten met deze veranderingen., Deze bevindingen voegen toe aan de huidige kennis van EBF3 gerelateerde aandoeningen en ondersteunen EBF3 haploinsufficižntie als sleutel in het neurodevelopmental syndrome geassocieerd met 10qter deleties.
materialen en methoden
de patiënt werd vastgesteld in het kader van een groot onderzoek naar neurologische ontwikkelingsstoornissen in Portugal, waarbij de registratie van de patiënten en families werd uitgevoerd door de verwijzende arts, klinische informatie werd verzameld in een anonieme database volgens de Portugese autoriteit voor gegevensbescherming (CNPD) en schriftelijke geïnformeerde toestemming werd verkregen voor alle deelnemers., Geïnformeerde toestemming voor de huidige patiënt werd verstrekt door de moeder voor de genetische studie en publicatie van de resultaten (inclusief foto ‘ s). De studie werd goedgekeurd door de ethische commissie van Center for Medical Genetics Dr.Jacinto Magalhães, National Health Institute Dr. Ricardo Jorge.
genomisch DNA werd geëxtraheerd uit perifeer bloed met behulp van een Citogene ® DNA-isolatiekit (Citomed, Portugal). aCGH werd uitgevoerd met Agilent 180 K array (AMADID:023363) tegen een diploïde DNA referentie (Kreatech ‘ s MegaPoll referentie DNA, Kreatech Diagnostics, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., De analyse werd uitgevoerd in een 7500-snelle realtime PCR-machine (Applied Biosystems, Foster City, CA, USA) met behulp van Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) volgens de aanbevelingen van de fabrikant en volgens de algemene richtlijnen voor qPCR. De specificiteit van elke reactie werd geverifieerd door de generatie van een smeltkromme voor elk van de versterkte fragmenten. De efficiëntie van de primer werd berekend door het genereren van een standaardcurve die past bij het geaccepteerde normale efficiëntiepercentage (gebruikte primers vermeld in aanvullende gegevens)., De Ct-waarden die voor elke test werden verkregen, werden geanalyseerd in dataassist™ – software (Applied Biosystems, Foster City, CA, USA).
resultaten
Hier beschrijven we een patiënt met een de novo deletie die EBF3 beïnvloedt. De patiënt is een 11-jarig meisje met ernstige ID (global development quotiënt = 27 op 7-jarige leeftijd), geboren uit niet-bloedverwante ouders en zonder familiegeschiedenis van neurologische ontwikkelingsstoornissen. Ze werd geboren na een biamniotische tweelingzwangerschap (haar zus is gezond), door vaginale bevalling, bij 35 weken zwangerschap. Geboorteparameters waren: gewicht, 1.830 g (P3); lengte, 42.,5 cm (P10); en OFC, 30,6 cm (P10), met een Apgar-score van 8/9 (respectievelijk 1e en 5e min). De neonatale periode werd gecompliceerd met sepsis en de diagnose van erfelijke sferocytose (geërfd van haar moeder). In de eerste maanden werd een globale ontwikkelingsachterstand geconstateerd, waarbij hoofdcontrole werd bereikt na 12 maanden, zitten op 18 maanden, zelfstandig lopen op 30 maanden en geen woorden werden gesproken op de leeftijd van 3 jaar., Ze had pyelonefritis op 19 maanden (renale echografie toonde geen afwijkingen), gastro-oesofageale reflux en terugkerende otitis media, met geleidend gehoorverlies dat chirurgische interventie en een gehoorapparaat vereist. Epilepsie werd vermoed na 5 maanden (episodes van onderbroken activiteit) maar het EEG was normaal.
ze werd voor het eerst waargenomen op de leeftijd van 3 jaar en 5 maanden (figuren 1A,B), toen vertoonde ze spierhypotonie, hypotoon gezicht, scheelzien en verminderde gevoeligheid voor pijn., Ze presenteerde ook milde dysmorfe kenmerken( figuur 1A): driehoekig gezicht, kleine lage oren met prominente anti-helix, gebogen wenkbrauwen, anteverted nares, bolvormige neuspunt, kleine mond met naar beneden gekeerde hoeken, spitse kin, korte nek, en prominente vinger foetale pads, evenals een milde korte gestalte (89 cm, overeenkomend met ongeveer 2SD).

figuur 1. (A) het gezicht van de patiënt na 3 jaar en 5 maanden met de kleine en lage oren met prominente anti-helix en (B) foetale pads in de vingers., (C) het gezicht van de patiënt op de leeftijd van 11 jaar. (D) gemarkeerd in grijs de 600 Kb verwijdering op 10q26.3 regio; een zoom in van het EBF3 gen in de DGV database onthult het bestaan van 3 verwijderingen in 3 controles die de eerste 6 exons van EBF3 beïnvloeden (NM_001005463); CNVs binnen deze regio gevonden in controle populaties omvatten verwijderingen nvs825626 (aanwezig in 1/31 individuen), nvs552315 (aanwezig in 1/17421 individuen) en nsv552316 (aanwezig in 1/31 individuen). (E) de schematische weergave van EBF3 transcripten.,
na 6 jaar werd een MRI van de hersenen uitgevoerd, maar er werden geen afwijkingen waargenomen. Op de leeftijd van 10 jaar werd ze opnieuw geëvalueerd; ze had nog steeds terugkerende otitis media, maar verder was in een goede wereldwijde gezondheid. De taal was zeer slecht (twee woordzinnen gesproken na 8 jaar). Ze had gedragsproblemen, met stereotiepe bewegingen (roterende bewegingen, kauwen op kleding, hoofd retropulsie), scoren voor ernstige autisme spectrum stoornis (ADI-R en ADOS) op 7 jaar; ze vertoonde agitatie en agressief gedrag (auto en hetero) en werd gemedicineerd met antipsychotica., Een orthopedische operatie werd uitgevoerd voor pes planus. De gelaatstrekken waren vergelijkbaar met die eerder beschreven, met afstand tussen de bovenste centrale snijtanden (figuur 1C); ze had brillen voor scheelzien en hypermetropie.
analyse van genomisch DNA door aCGH toonde een de novo 600 kb deletie aan bij 10p26. 3 (figuur 1D) die drie genen aantast—MGMT (codeert het enzym o-6-methylguanine-DNA methyltransferase, betrokken bij DNA-herstel), EBF3 en GLRX (codeert glutaredoxine, een kleine thioltransferase die eiwit-GSH-adducten verwijdert), waarvan EBF3 het meest waarschijnlijke ziektegeassocieerde gen was.,
discussie
de gepresenteerde patiënt werd enkele jaren geleden voor het eerst geanalyseerd door aCGH. Op het moment van acgh analyse, het bestaan van de drie varianten aanwezig in Database van Genomic varianten (DGV) 1 die de eerste vijf exons van EBF3 gen (figuur 1E; Park et al., 2010; Cooper et al., 2011), evenals de afwezigheid van andere bekende ziekte veroorzakende mutaties in dit gen, leiden ons om het te classificeren als een variant van onbekende betekenis (VOUS). Echter, de recente publicaties van EBF3 mutaties (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) en de klinische overeenkomsten met de gerapporteerde gevallen, leiden ons om de variant opnieuw te beoordelen en ons laten geloven dat EBF3-verwijdering in feite kan worden goed voor de ziekte in de patiënt. Een van de aspecten die twijfels oproept over de pathogeniteit van deze variant in de eerste plaats was het bestaan van populatiecontroles met verwijderingen van de eerste zes exonen van dit gen, in heterozygositeit (gegevens uit de DGV-database vanaf februari 2017) (figuur 1D). Hoewel een transcript van EBF3 beginnend in Exon13b is opgenomen in de Ensembl database (ENST00000440978.,1) (Figuur 1E), die zou kunnen verklaren hoe verwijdering van de eerste exons uiteindelijk zou kunnen resulteren in een normaal fenotype, dit transcript sluit het DNA-bindingsdomein van EBF3 uit, en zijn expressiepatroon en functionele relevantie zijn niet gekarakteriseerd. Na herbeoordeling, echter, de CNV ’s beschreven door Cooper en collega’ s (nsv552315 en nsv552316) (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) werden beschouwd als op de drempel van detectie door SNP microarray en kan niet de basis zijn voor uitsluiting van een kandidaat-gen, in het bijzonder in het licht van het sterke genetische en functionele bewijs voor de relevantie van EBF3-mutaties voor de ziekte (Evan Eichler, Greg Cooper en Bradley Coe, persoonlijke communicatie).,
onze patiënt vertoont veel klinische overeenkomsten met eerder beschreven patiënten met mutaties in EBF3 (21 gevallen samengevat in Tabel 1), zoals globale ontwikkelingsachterstand, vertraagde expressieve spraak, hypotonie, verhoogde pijndrempel, gedragsproblemen en karakteristieke gelaatstrekken (lang/driehoekig gezicht, groot voorhoofd, hypotonisch gezicht)., Nochtans, hoewel onze patiënt significante vertraging in motorische ontwikkeling had, werd geen significante ataxie ontdekt (met sommige beperkingen in klinisch onderzoek, aangezien het kind niet coöperatief was), en geen cerebellaire anomalieën waren aanwezig in hersenen MRI.

Tabel 1. Klinische vergelijking van het onderhavige geval met de gemelde gevallen met puntmutaties/indels in het ebf3-gen.
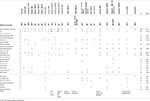
Tabel 2., Klinische vergelijking van het onderhavige geval met de gemelde gevallen met verwijderingen van de 10q26 cytoband.
De EBF3-varianten die begin 2017 in de literatuur zijn beschreven, omvatten puntmutaties waarvan wordt verwacht dat ze schadelijk zijn en kleine inserties en deleties die leiden tot in frame deletie van belangrijke aminozuren of tot een frameverschuiving, die voorspelbaar vroege afkaping van het resulterende eiwit of Nonsens-gemedieerd verval veroorzaken., De mutaties waren geconcentreerd op Delen van het gen dat codeert voor het DNA-bindende domein van EBF3, en werden voorspeld door verschillende methoden om te leiden tot een verlies van functie van deze transcriptiefactor, waardoor verminderde functie en haploinsufficiëntie worden voorgesteld als het mechanisme dat ten grondslag ligt aan de neurologische ontwikkelingsstoornis bij deze patiënten. Knock-out Muizen Voor Ebf3 worden beschreven om neonatale letaliteit en neuronale migratie defecten te presenteren, met falen van olfactorische neuronen om te projecteren naar de dorsale olfactorische bol (Wang et al.,, 2004), maar er wordt geen beschrijving gegeven van een fenotype bij heterozygote dieren, die in veel van de experimenten als controles worden gepresenteerd, waardoor het haploinsufficiëntiemodel niet wordt ondersteund. We hebben ons ingespannen om het neurologische fenotype van deze dieren te verkrijgen en te bestuderen, maar waren niet succesvol, omdat de Ebf3(O/E2) knock-out lijn Misschien is stopgezet (Joseph W. Lewcock, personal communication). Echter, het huidige geval samen met de patiënten samengevat in Tabel 2, ondersteunen de hypothese van EBF3 haploinsufficiency als ziekte veroorzakend.,
samengevat versterkt de huidige beschrijving EBF3-functieverlies/haploinsufficiëntie als oorzaak van neurodevelopmentale ziekte, en versterkt de associatie van dit gen met een karakteristiek klinisch syndroom binnen dit spectrum.
Auteursbijdragen
FL voerde de moleculaire studies uit en analyseerde de moleculaire gegevens. MG en GS verzamelden klinische gegevens. FL, JP en PM hebben alle gevallen van EBF3-mutatie in de literatuur beoordeeld. FL, GS, JP en PM hebben de paper opgesteld. PM kreeg financiering voor deze studie. De studie werd uitgevoerd onder leiding van PM.,
financiering
FCT-Fundação para a Ciência e a Tecnologia binnen de projecten en beurzen (PIC/IC/83026 / 2007, PIC/IC/83013/2007, SFRH/BD/90167 / 2012). Dit artikel is ontwikkeld in het kader van het project NORTE-01-0145-FEDER-000013, ondersteund door het regionaal operationeel programma voor Noord-Portugal (NORTE 2020), in het kader van de Partnerschapsovereenkomst Portugal 2020, via het Europees Fonds voor Regionale Ontwikkeling (FEDER).
Conflict of Interest Statement
JP werd gebruikt door het bedrijf CGC Genetics.,
de andere auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
erkenningen
we willen de patiënt en haar familie bedanken voor hun deelname aan de genetische studies en voor het toestaan van deze publicatie. We zijn Dr. Ana Maria Fortuna en Dr. Margarida Reis Lima dankbaar voor het mogelijk maken van dit project en het faciliteren van onze samenwerking met CGM.
voetnoten
1., ^Database van genomische varianten beschikbaar op: http://dgv.tcag.ca/dgv/app/home .
Friocourt, G., and Parnavelas, J. (2011). Identificatie van Arx targets onthult nieuwe kandidaten voor het beheersen van corticale interneuron migratie en differentiatie. Voorkant. Cel. Neurowetenschappen. 5:28. doi: 10.3389 / fncel.2011.00028
PubMed Abstract / CrossRef Full Text / Google Scholar
















Geef een reactie