syndrome de Turner masculin, syndrome de Noonan-Ehmke, syndrome de Turner-like, syndrome D’Ullrich-Noonan
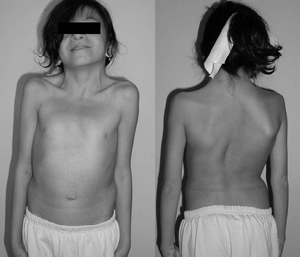
Une fille de 12 ans atteinte du syndrome de Noonan. Cou palmé typique. Double courbe structurelle avec déformation des côtes.,irmed avec tests génétiques
syndrome Cardiofaciocutané, syndrome de Turner, syndrome de Costello, neurofibromatose de type 1
basé sur les symptômes
hormone de croissance
dépend de la gravité des problèmes cardiaques
1 sur 100 (1 maladie grave sur 2 000)
Le syndrome de Noonan (NS) est une maladie génétique qui peut présenter des traits du visage légèrement inhabituels, une taille courte, une cardiopathie congénitale, des problèmes de saignement et des malformations squelettiques., Les traits du visage comprennent des yeux largement espacés, des yeux de couleur claire, des oreilles basses, un cou court et une petite mâchoire inférieure. Les problèmes cardiaques peuvent inclure une sténose valvulaire pulmonaire. L’os du sein peut faire saillie ou être coulé, tandis que la colonne vertébrale peut être anormalement courbée. L’Intelligence dans le syndrome est souvent normale. Les Complications de la NS peuvent inclure la leucémie.
un certain nombre de mutations génétiques peuvent entraîner le syndrome de Noonan. La condition peut être héritée des parents d’une personne comme une condition autosomique dominante ou se produire comme une nouvelle mutation., Le syndrome de Noonan est un type de Rasopathie, dont le mécanisme sous-jacent implique une suractivation dans la voie de signalisation cellulaire RAS/MAPK. Le diagnostic peut être suspecté en fonction des symptômes, de l’imagerie médicale et des tests sanguins. La Confirmation peut être obtenue par des tests génétiques.
aucun remède pour la NS n’est connu. Le traitement est basé sur les symptômes et les problèmes sous-jacents, et un soutien supplémentaire à l’école peut être nécessaire. L’hormonothérapie de croissance pendant l’enfance peut augmenter la taille finale d’une personne affectée. Les résultats à Long terme dépendent généralement de la gravité des problèmes cardiaques.,
on estime que 1 personne sur 1 000 est légèrement touchée par la NS, tandis qu’environ 1 personne sur 2 000 a une forme plus grave de la maladie. Les mâles semblent être touchés plus souvent que les femelles. La condition a été décrite pour la première fois en 1883 et a été nommée d’après la cardiologue pédiatrique américaine Jacqueline Noonan, qui a décrit d’autres cas en 1963.
signes et symptômes

caractéristiques anormales du syndrome de Noonan à l’âge de 3 mois: notez l’inclinaison des sourcils et la chute de la paupière gauche.,

Anormale des caractéristiques du syndrome de Noonan, à l’âge de 3 mois: Notez le bas, en arrière de rotation, et anormalement formé l’oreille.
Les signes les plus courants conduisant au diagnostic du syndrome de Noonan sont des caractéristiques faciales uniques et des caractéristiques musculo-squelettiques. Les caractéristiques faciales sont les plus importantes dans la petite enfance, devenant moins apparentes avec l’âge chez de nombreuses personnes atteintes du syndrome de Noonan.,
tête
certaines des caractéristiques du syndrome de Noonan comprennent une grande tête avec un excès de peau à l’arrière du cou, une racine des cheveux basse à la nuque, une racine des cheveux haute à l’avant de la tête, une forme de visage triangulaire, un front large et un cou palmé court.
dans les yeux, l’hypertélorisme (yeux largement fixés) est une caractéristique déterminante, présente chez 95% des personnes atteintes du syndrome de Noonan., Cela peut s’accompagner de plis épicanthaux (pli supplémentaire de la peau au coin interne de l’Œil), de ptosis (affaissement des paupières), de proptosis (yeux exorbités), de strabisme (retournement des yeux vers l’intérieur ou vers l’extérieur), de nystagmus (mouvement saccadé des yeux) et d’erreurs visuelles réfractives.
le nez peut être petit, large et retourné.
le Développement de la bouche peut également être affecté dans le syndrome de Noonan., Cela peut entraîner un philtrum profondément rainuré (ligne de la lèvre supérieure) (plus de 90%), une micrognathie (mâchoire inférieure sous-dimensionnée), un palais arqué élevé, des difficultés d’articulation (les dents ne s’alignent pas) qui peuvent entraîner des problèmes dentaires. Semblable aux manifestations musculaires ci-dessus, dans la bouche, un mauvais contrôle de la langue peut être observé.,
peau
Les signes et symptômes cutanés du syndrome de Noonan comprennent le lymphœdème (gonflement lymphatique des extrémités), la formation de chéloïdes, la formation excessive de cicatrices, l’hyperkératose (surdéveloppement de la couche externe de la peau), les naevus pigmentés (taches cutanées foncées) et la maladie du tissu conjonctif
musculo-squelettique
des anomalies des membres et des extrémités peuvent survenir dans le syndrome de Noonan. Cela peut se manifester par des doigts émoussés, un rembourrage supplémentaire sur les doigts et les orteils, un œdème du dos des mains et du dessus des pieds et un cubitus valgus (grand angle de transport des coudes).,
pour la petite taille, l’hormone de croissance est parfois combinée avec IGF-1 (ou comme alternative, IGF-1 comme autonome) peut être employée pour réaliser une taille accrue / taille finale plus rapidement. La taille adulte finale des individus atteints du syndrome de Noonan est d’environ 161-167 cm chez les mâles et 150-155 cm chez les femelles, ce qui s’approche de la limite inférieure de la normale.
Épinière anomalies peuvent être présentes jusqu’à 30% du temps, et cela peut nécessiter une intervention chirurgicale pour corriger dans plus de 60% de ces cas., D’autres manifestations musculo-squelettiques du syndrome de Noonan sont associées à des troubles du tissu conjonctif indifférenciés pouvant être associés à des contractures articulaires (oppression) ou à une hypermobilité articulaire (relâchement). D’autres facteurs peuvent se présenter sous la forme de l’aile de l’omoplate, de la scoliose, de la proéminence de l’os du sein (pectus carinatum), de la dépression de l’os du sein (pectus excavatum). Les anomalies musculaires peuvent se présenter sous forme d’hypotonie (faible tonus musculaire), ce qui peut entraîner une lordose (augmentation du creux dans le dos) en raison d’un mauvais tonus musculaire abdominal.,
coeur
le syndrome de Noonan est la deuxième cause syndromique la plus fréquente de cardiopathie congénitale. Cela comprend la sténose valvulaire pulmonaire (50-60%), les défauts septaux auriculaires (10-25%), les défauts septaux ventriculaires (5-20%) et la cardiomyopathie hypertrophique (12-35%).
poumons
Une fonction pulmonaire Restrictive a été rapportée chez certaines personnes.
gastro-intestinal
un certain nombre de divers symptômes gastro-intestinaux (IG) ont été associés au syndrome de Noonan., Ceux-ci comprennent des difficultés à avaler, une faible motilité intestinale, une gastroparésie (vidange gastrique retardée), une malrotation intestinale et des vomissements fréquents ou violents. Ces problèmes digestifs peuvent entraîner une diminution de l’appétit, une incapacité à prospérer de la petite enfance à la puberté (75%) et parfois le besoin d’une sonde d’alimentation.{ Il a été associé à la maladie de Von Willebrand, à une thrombocytopénie Amégakaryocytaire (faible numération plaquettaire), à un temps prolongé de thromboplastine partielle activée, à des défauts de coagulation combinés., Lorsqu’ils sont présents, ces troubles d’accompagnement du syndrome de Noonan peuvent être associés à une prédisposition à une ecchymose facilement ou à une hémorragie.
neurologique
parfois, une malformation de Chiari (type 1) peut survenir,ce qui peut entraîner une hydrocéphalie. Des saisies ont également été signalées.
Cause

NS est généralement transmis selon un mode autosomique dominant avec une expression variable.,
la récurrence chez les frères et sœurs et la transmission apparente du parent à l’enfant ont longtemps suggéré un défaut génétique avec un héritage autosomique dominant et une expression variable. On sait que les Mutations dans les voies de signalisation de la protéine kinase activée par Ras/mitogène sont responsables d’environ 70% des cas de NS.
Les personnes atteintes de NS ont jusqu’à 50% de chances de le transmettre à leur progéniture., Le fait qu’un parent affecté ne soit pas toujours identifié pour les enfants atteints de NS suggère plusieurs possibilités:
- Les Manifestations pourraient être si subtiles qu’elles ne seraient pas reconnues (expressivité variable)
- NS est hétérogène, comprenant plus d’une condition similaire de causes différentes, et certaines d’entre elles peuvent ne pas être héritées.
- Une forte proportion de cas peut représenter de nouvelles, mutations sporadiques.,
| Type | en Ligne Hérédité Mendélienne à l’Homme de base de données | Gène | Année | Locus | % des cas | Description | Réf. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | Le gène PTPN11 code pour la protéine tyrosine phosphatase SHP-2., Cette protéine est un composant de plusieurs voies de transduction de signal intracellulaire impliquées dans le développement embryonnaire qui modulent la division cellulaire, la différenciation et la migration, y compris l’une médiée par le récepteur du facteur de croissance épidermique, qui est important dans la formation des valves cardiaques semi-lunaires. La Duplication de la région chromosomique contenant PTPN11 peut également entraîner la NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
des mutations hétérozygotes dans les ARN, ARH, BRAF, shoc2, map2k1, map2k2 et CBL ont également été associées à un pourcentage plus faible de ns et de phénotypes apparentés.
une affection connue sous le nom de « syndrome de neurofibromatose-Noonan » est associée à la neurofibromine.
diagnostic
La NS peut être confirmée génétiquement par la présence de l’une des mutations connues énumérées ci-dessus., Cependant, malgré l’identification de 14 gènes responsables, l’absence d’une mutation connue n’exclura pas le diagnostic, car d’autres gènes encore non découverts peuvent causer la NS. Ainsi, le diagnostic de NS est toujours basé sur des caractéristiques cliniques. En d’autres termes, il est fait quand un médecin estime qu’une personne a suffisamment de fonctionnalités pour justifier l’étiquette. Les principales valeurs d’un diagnostic génétique sont qu’il guide des évaluations médicales et développementales supplémentaires, qu’il exclut d’autres explications possibles pour les caractéristiques et qu’il permet des estimations plus précises du risque de récidive., Avec plus d’études de corrélation génotype-phénotype en cours, un diagnostic génétique positif aidera le clinicien à être conscient des anomalies possibles spécifiques à cette mutation génétique. Par exemple, une augmentation de la cardiomyopathie hypertrophique est observée chez les personnes présentant une mutation de KRAS et un risque accru de leucémie myélomonocytaire juvénile existe pour une mutation de PTPN11. À l’avenir, des études pourraient conduire à une prise en charge ciblée des symptômes de la NS qui dépend de la mutation génétique d’une personne.,
avant la naissance
Les caractéristiques prénatales qui pourraient amener les médecins à envisager un diagnostic de syndrome de Noonan comprennent l’hygroma kystique, l’augmentation de la translucidité nucale, l’épanchement pleural et l’œdème.
diagnostic différentiel
bien que le syndrome de Turner présente des similitudes avec les anomalies rénales et le retard de développement, le syndrome de Turner ne se trouve que chez les femmes et s’exprime souvent différemment. Dans le syndrome de Turner, il y a une incidence plus faible de retards de développement, les malformations cardiaques du côté gauche sont constantes et l’apparition d’anomalies rénales est beaucoup plus faible.,
autres RASopathies
- syndrome de Watson – le Syndrome de Watson présente un certain nombre de caractéristiques similaires au Syndrome de Noonan, telles que la petite taille, la sténose valvulaire pulmonaire, le développement intellectuel variable et les changements de pigment cutané.
- syndrome Cardiofaciocutané (CFC) – le syndrome CFC est très similaire au Syndrome de Noonan en raison de caractéristiques cardiaques et lymphatiques similaires. Cependant, dans le syndrome CFC, la déficience intellectuelle et les problèmes gastro-intestinaux sont souvent plus graves et prononcés.,
- syndrome de Costello – comme le syndrome CFC, le syndrome de Costello a des caractéristiques qui se chevauchent avec le Syndrome de Noonan. Cependant, les conditions peuvent être distinguées par leur cause génétique.
- neurofibromatose 1 (NF1)
- syndrome de Williams
prise en charge
le traitement varie en fonction des complications mais tend à être assez standard, reflétant le traitement de la population générale., Des directives de prise en charge, divisées par systèmes, y compris général, développemental, dentaire, croissance et alimentation, cardiovasculaire, audiologique, hématologique, rénal et squelettique, qui rendent compte des actions à prendre au diagnostic, après le diagnostic et si symptomatique, ont été publiées par un consortium américain.
plus précisément, le traitement des complications cardiovasculaires ressemble à celui de la population générale et le traitement de la diathèse hémorragique est guidé par le déficit en facteur spécifique ou l’agrégation plaquettaire.,
- Les tests neuropsychologiques sont recommandés pour trouver les forces et les défis afin d’adapter le soutien nécessaire à l’école et à la carrière.
- Une personnalisation de L’éducation telle qu’un plan de programme d’éducation individualisé est parfois nécessaire pour les enfants d’âge scolaire.
- orthophonie en présence de problèmes d’élocution et d’articulation
- physiothérapie et ergothérapie pour les retards moteurs grossiers et fins
- L’hypotonie et les difficultés motrices ont souvent un impact sur l’écriture. Les aménagements pour réduire les demandes d’écriture manuscrite amélioreront les performances et économiseront la fonction manuelle à long terme.,
- Un suivi périodique et une surveillance à vie des anomalies observées dans n’importe quel système, en particulier le système cardiovasculaire, sont recommandés.
risque D’anesthésie
bien que quelques personnes atteintes du syndrome de Noonan aient été signalées pour développer une hyperthermie maligne, la mutation génétique des maladies connues pour être associées à une hyperthermie maligne est différente de celle du syndrome de Noonan.,
pronostic
la durée de vie des personnes atteintes du syndrome de Noonan peut être similaire à celle de la population générale, cependant, le syndrome de Noonan peut être associé à plusieurs problèmes de santé pouvant contribuer à la mortalité. Les complications des maladies cardiovasculaires sont le principal facteur de mortalité chez les personnes atteintes du syndrome de Noonan. Le pronostic dépend donc largement de la présence ou de l’absence de maladie cardiaque, ainsi que du type et de la gravité de la maladie (si la maladie est présente)., Plus particulièrement, le syndrome de Noonan avec cardiomyopathie hypertrophique est associé à une mortalité accrue.
histoire
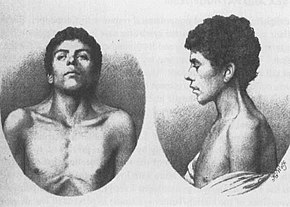
le plus ancien cas connu de NS, décrit en 1883 par Kobylinski
Jacqueline Noonan exerçait en tant que cardiologue pédiatrique à l’Université de Iowa quand elle a remarqué que les enfants atteints d’un type rare de malformation cardiaque, la sténose pulmonaire valvulaire, avaient souvent une apparence physique caractéristique, avec une petite taille, un cou palmé, de grands yeux espacés et des oreilles basses., Les garçons et les filles ont été touchés. Ces caractéristiques ont parfois été observées dans les familles, mais n’étaient pas associées à des anomalies chromosomiques grossières. Elle a étudié 833 personnes atteintes du syndrome de Noonan à la clinique des cardiopathies congénitales, à la recherche d’autres anomalies congénitales, et en 1963 a présenté un article: « malformations non cardiaques associées chez les enfants atteints de cardiopathie congénitale ». Cela a décrit 9 enfants qui, en plus des cardiopathies congénitales, présentaient des traits faciaux caractéristiques, des déformations thoraciques et une petite taille.
Dr. John Opitz, un ancien élève du Dr., Noonan, a commencé à appeler la condition « syndrome de Noonan » quand il a vu des enfants qui ressemblaient à ceux que le Dr Noonan avait décrits. La Dre Noonan a produit un article intitulé « Hypertélorism with Turner Phenotype » en 1968 où elle a étudié 19 patients qui présentaient des symptômes indicatifs du Syndrome de Noonan. En 1971, lors du Symposium sur les malformations cardiovasculaires, le nom « syndrome de Noonan » a été officiellement reconnu.
















Laisser un commentaire