Maschio di Turner sindrome di Noonan-Ehmke, la sindrome di Turner, sindrome simil -, Ullrich-la sindrome di Noonan
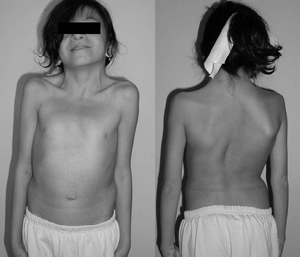
Un 12-anno-vecchia ragazza con la sindrome di Noonan. Tipico collo palmato. Doppia curva strutturale con deformità costale.,irmed con i test genetici
Cardiofaciocutaneous, sindrome di, sindrome di Turner, sindrome di Costello, la neurofibromatosi di tipo 1
sulla Base dei sintomi
l’ormone della Crescita
Dipende dalla gravità dei problemi di cuore
1 a 100 (1 caso su 2000 grave malattia)
la sindrome di Noonan (NS) è una malattia genetica che si possono presentare con un eufemismo caratteristiche facciali inusuali, a breve altezza, cardiopatie congenite, problemi di sanguinamento, e malformazioni scheletriche., Le caratteristiche facciali includono occhi ampiamente distanziati, occhi di colore chiaro, orecchie basse, un collo corto e una piccola mascella inferiore. I problemi cardiaci possono includere stenosi della valvola polmonare. L’osso del seno può sporgere o essere affondata, mentre la colonna vertebrale può essere anormalmente curvo. L’intelligenza nella sindrome è spesso normale. Le complicanze di NS possono includere la leucemia.
Un certo numero di mutazioni genetiche può causare la sindrome di Noonan. La circostanza può essere ereditata dai genitori di una persona come circostanza dominante autosomica o accadere come nuova mutazione., La sindrome di Noonan è un tipo di RASopatia, il cui meccanismo sottostante comporta l’iperattivazione all’interno della via di segnalazione cellulare RAS / MAPK. La diagnosi può essere sospettata sulla base di sintomi, imaging medico e esami del sangue. La conferma può essere ottenuta con test genetici.
Non è nota alcuna cura per NS. Il trattamento si basa sui sintomi e sui problemi sottostanti e potrebbe essere richiesto un supporto extra a scuola. La terapia dell’ormone della crescita durante l’infanzia può aumentare l’altezza finale di una persona colpita. I risultati a lungo termine dipendono in genere dalla gravità dei problemi cardiaci.,
Si stima che 1 persona su 1000 sia lievemente affetta da NS, mentre circa 1 su 2.000 ha una forma più grave della condizione. I maschi sembrano essere colpiti più spesso delle femmine. La condizione fu descritta per la prima volta nel 1883 e prese il nome dal cardiologo pediatrico americano Jacqueline Noonan, che descrisse ulteriori casi nel 1963.
Segni e sintomi

Caratteristiche anormali della sindrome di Noonan all’età di 3 mesi: notare l’inclinazione del sopracciglio e la caduta della palpebra sinistra.,

Caratteristiche anormali della sindrome di Noonan all’età di 3 mesi: si noti l’orecchio impostato in basso, ruotato posteriormente e formato in modo anomalo.
I segni più comuni che portano alla diagnosi della sindrome di Noonan sono caratteristiche facciali uniche e caratteristiche muscolo-scheletriche. Le caratteristiche facciali sono più importanti nell’infanzia, diventando meno evidenti con l’età in molte persone con sindrome di Noonan.,
Testa
Alcune delle caratteristiche della sindrome di Noonan includono una grande testa con pelle in eccesso sulla parte posteriore del collo, bassa attaccatura dei capelli alla nuca, alta attaccatura dei capelli nella parte anteriore della testa, forma triangolare del viso, fronte ampia e un collo corto e palmato.
Negli occhi, l’ipertelorismo (occhi ampiamente impostati) è una caratteristica distintiva, presente nel 95% delle persone con sindrome di Noonan., Questo può essere accompagnato da pieghe epicantali (piega extra della pelle all’angolo interno dell’occhio), ptosi (abbassamento delle palpebre), proptosi (occhi sporgenti), strabismo (rotazione verso l’interno o verso l’esterno degli occhi), nistagmo (movimento a scatti degli occhi) e errori visivi rifrattivi.
Il naso può essere piccolo, largo e rovesciato.
Lo sviluppo della bocca può anche essere influenzato dalla sindrome di Noonan., Ciò può provocare philtrum profondamente scanalato (linea superiore del labbro) (oltre il 90%), micrognazia (mascella inferiore sottodimensionata), palato ad arco alto, difficoltà di articolazione (i denti non si allineano) che possono portare a problemi dentali. Simile alle manifestazioni muscolari di cui sopra, in bocca, si può osservare uno scarso controllo della lingua.,
Pelle
Pelle segni e sintomi nella sindrome di Noonan includono linfedema (linfatici gonfiore delle estremità), formazione di cheloidi, eccessiva formazione di cicatrice, ipercheratosi (eccessivo sviluppo di strato cutaneo esterno), nevi pigmentati (pigmentazione scura punti), e connettivo malattie del tessuto
muscolo-scheletrico
Anomalie degli arti e delle arti, può verificarsi la sindrome di Noonan. Questo può manifestarsi come dita senza mezzi termini, imbottitura extra sulle dita delle mani e dei piedi, edema del dorso delle mani e della parte superiore dei piedi e cubito valgo (ampio angolo di trasporto dei gomiti).,
Per bassa statura, l’ormone della crescita è talvolta combinato con IGF-1 (o in alternativa, IGF-1 come stand-alone) può essere utilizzato per ottenere un’altezza maggiore/altezza finale più veloce. L’altezza adulta finale degli individui con sindrome di Noonan è di circa 161-167 cm nei maschi e 150-155 cm nelle femmine, che si avvicina al limite inferiore del normale.
Anomalie spinali possono essere presenti fino al 30% del tempo e questo può richiedere un intervento chirurgico per correggere in oltre il 60% di questi casi., Altre manifestazioni muscoloscheletriche nella sindrome di Noonan sono associate a disturbi indifferenziati del tessuto connettivo che possono essere associati a contratture articolari (oppressione) o ipermobilità articolare (scioltezza). Ulteriori fattori possono presentare sotto forma di ali della scapola, scoliosi, prominenza dell’osso del seno (pectus carinatum), depressione dell’osso del seno (pectus excavatum). Anomalie muscolari possono presentarsi come ipotonia (basso tono muscolare), che può portare a lordosi (aumento della cavità nella parte posteriore) a causa di scarso tono muscolare addominale.,
Cuore
La sindrome di Noonan è la seconda causa sindromica più comune di cardiopatia congenita. Ciò include stenosi valvolare polmonare (50-60%), difetti del setto atriale (10-25%), difetti del setto ventricolare (5-20%) e cardiomiopatia ipertrofica (12-35%).
Polmoni
In alcune persone è stata riportata una funzione polmonare restrittiva.
Gastrointestinale
Un certo numero di diversi sintomi gastrointestinali (GI) sono stati associati alla sindrome di Noonan., Questi includono difficoltà di deglutizione, bassa motilità intestinale, gastroparesi (svuotamento gastrico ritardato), malrotazione intestinale e vomito frequente o forzato. Questi problemi digestivi possono portare a diminuzione dell’appetito, incapacità di prosperare dall’infanzia alla pubertà (75%) e, occasionalmente, la necessità di un tubo di alimentazione.{ È stato associato alla malattia di Von Willebrand, trombocitopenia amegacariocitica (bassa conta piastrinica), tempo prolungato di tromboplastina parziale attivata, difetti di coagulazione combinati., Quando presente, questi disturbi di accompagnamento della sindrome di Noonan possono essere associati a una predisposizione a lividi facilmente o emorragia.
Neurologico
Occasionalmente, può verificarsi una malformazione di Chiari (tipo 1),che può portare all’idrocefalo. Sono state riportate anche convulsioni.
Cause

NS è tipicamente ereditato in un modello autosomico dominante con espressione variabile.,
La ricorrenza nei fratelli e la trasmissione apparente da genitore a figlio hanno suggerito a lungo un difetto genetico con ereditarietà autosomica dominante e espressione variabile. Le mutazioni nelle vie di segnalazione delle protein chinasi attivate da Ras / mitogen sono note per essere responsabili di circa il 70% dei casi di NS.
Le persone con NS hanno fino al 50% di possibilità di trasmetterlo alla loro prole., Il fatto che un genitore affetto non sia sempre identificato per i bambini con NS suggerisce diverse possibilità:
- Le manifestazioni potrebbero essere così sottili da non essere riconosciute (espressività variabile)
- NS è eterogeneo, comprendente più di una condizione simile di cause diverse, e alcune di queste potrebbero non essere ereditate.
- Un’alta percentuale di casi può rappresentare nuove mutazioni sporadiche.,
| Tipo | Online Ereditarietà Mendeliana nell’Uomo il database | Gene | Anno trovato | Locus | % di casi | Descrizione | Rif. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | Il gene PTPN11 codifica la proteina tirosina fosfatasi SHP-2., Questa proteina è un componente di diverse vie di trasduzione del segnale intracellulare coinvolte nello sviluppo embrionale che modulano la divisione cellulare, la differenziazione e la migrazione, inclusa una mediata dal recettore del fattore di crescita epidermico, che è importante nella formazione delle valvole cardiache semilunari. La duplicazione della regione cromosomica contenente PTPN11 può anche causare NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3 p 25 | 3-17% |
mutazioni Eterozigoti ANR, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, e CBL sono stati anche associati con una minore percentuale di NS e fenotipi correlati.
Una condizione nota come “sindrome di neurofibromatosi-Noonan” è associata alla neurofibromina.
Diagnosi
NS può essere confermata geneticamente dalla presenza di una qualsiasi delle mutazioni note sopra elencate., Tuttavia, nonostante l’identificazione di 14 geni causali, l’assenza di una mutazione nota non escluderà la diagnosi, poiché più geni non ancora scoperti possono causare NS. Pertanto, la diagnosi di NS si basa ancora su caratteristiche cliniche. In altre parole, è fatto quando un medico sente che una persona ha abbastanza delle caratteristiche per giustificare l’etichetta. I valori principali di fare una diagnosi genetica sono che guida ulteriori valutazioni mediche e dello sviluppo, esclude altre possibili spiegazioni per le caratteristiche e consente stime del rischio di recidiva più accurate., Con più studi di correlazione genotipo-fenotipo in corso, una diagnosi genetica positiva aiuterà il clinico ad essere a conoscenza di possibili anomalie specifiche di quella determinata mutazione genica. Ad esempio, un aumento della cardiomiopatia ipertrofica è visto nelle persone con una mutazione di KRAS e un aumento del rischio di leucemia mielomonocitica giovanile esiste per una mutazione di PTPN11. In futuro, gli studi possono portare a una gestione mirata dei sintomi di NS che dipende da quale mutazione genetica ha una persona.,
Prima della nascita
Le caratteristiche prenatali che potrebbero portare i medici a considerare una diagnosi di sindrome di Noonan includono l’igroma cistico, l’aumento della traslucenza nucale, il versamento pleurico e l’edema.
Diagnosi differenziale
Mentre la sindrome di Turner ha somiglianze con anomalie renali e ritardo dello sviluppo, la sindrome di Turner si trova solo nelle femmine e spesso esprime in modo diverso. Nella sindrome di Turner, vi è una minore incidenza di ritardi nello sviluppo, i difetti cardiaci del lato sinistro sono costanti e l’insorgenza di anomalie renali è molto più bassa.,
Altre rasopatie
- Sindrome di Watson – La sindrome di Watson ha una serie di caratteristiche simili con la sindrome di Noonan come bassa statura, stenosi della valvola polmonare, sviluppo intellettuale variabile e cambiamenti del pigmento cutaneo.
- Sindrome cardiofaciocutanea (CFC) – La sindrome CFC è molto simile alla sindrome di Noonan a causa di caratteristiche cardiache e linfatiche simili. Tuttavia, nella sindrome da CFC la disabilità intellettiva e i problemi gastrointestinali sono spesso più gravi e pronunciati.,
- Sindrome di Costello – Come la sindrome CFC, la sindrome di Costello ha caratteristiche sovrapposte con la sindrome di Noonan. Tuttavia, le condizioni possono essere distinte dalla loro causa genetica.
- Neurofibromatosi 1 (NF1)
- Sindrome di Williams
Gestione
Il trattamento varia a seconda delle complicanze ma tende ad essere abbastanza standard, riflettendo il trattamento della popolazione generale., Le linee guida di gestione, suddivise per sistemi, tra cui generale, dello sviluppo, dentale, crescita e alimentazione, cardiovascolare, audiologico, ematologico, renale e scheletrico, che tengono conto delle azioni da intraprendere alla diagnosi, dopo la diagnosi e se sintomatica, sono state pubblicate da un consorzio americano.
In particolare, il trattamento delle complicanze cardiovascolari assomiglia a quello della popolazione generale e il trattamento della diatesi emorragica è guidato dalla carenza di fattori specifici o dall’aggregazione piastrinica.,
- Test neuropsicologici si consiglia di trovare i punti di forza e le sfide per adattare il supporto necessario per la scuola e la carriera.
- Personalizzazione educativa come un piano di programma di educazione individualizzato a volte è necessario per i bambini in età scolare.
- Logopedia se si presentano problemi di linguaggio e articolazione
- Terapia fisica e terapia occupazionale per ritardi motori grossolani e fini
- L’ipotonia e le difficoltà motorie hanno spesso un impatto sulla scrittura a mano. Le sistemazioni per ridurre le richieste di scrittura a mano miglioreranno le prestazioni e risparmieranno la funzione della mano a lungo termine.,
- Si raccomanda un follow-up periodico e un monitoraggio permanente delle anomalie riscontrate in qualsiasi sistema, in particolare nel sistema cardiovascolare.
Rischio di anestesia
Sebbene alcune persone con sindrome di Noonan siano state segnalate per sviluppare ipertermia maligna, la mutazione genetica delle malattie note per essere associate all’ipertermia maligna è diversa da quella della sindrome di Noonan.,
Prognosi
La durata della vita delle persone con sindrome di Noonan può essere simile alla popolazione generale, tuttavia, la sindrome di Noonan può essere associata a diverse condizioni di salute che possono contribuire alla mortalità. Il più grande contributore alla mortalità in individui con sindrome di Noonan è complicazioni della malattia cardiovascolare. La prognosi dipende quindi in gran parte dalla presenza o dall’assenza di malattie cardiache, nonché dal tipo e dalla gravità della malattia (se la malattia è presente)., In particolare, la sindrome di Noonan con cardiomiopatia ipertrofica è associata ad un aumento della mortalità.
Storia
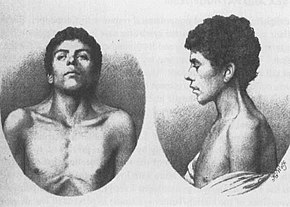
Il più antico caso di NS, descritto nel 1883 da Kobylinski
Jacqueline Noonan stato in pratica come un cardiologo pediatrico presso l’Università di Iowa, quando ha notato che i bambini con un raro tipo di difetto cardiaco, valvolare stenosi polmonare, spesso con un caratteristico aspetto fisico, con bassa statura, pterigio del collo, distanziate occhi, e basse e le orecchie., Sia i ragazzi che le ragazze sono stati colpiti. Queste caratteristiche sono state a volte visto in esecuzione in famiglie, ma non sono stati associati con anomalie cromosomiche grossolane. Ha studiato 833 persone con sindrome di Noonan presso la clinica di cardiopatia congenita, alla ricerca di altre anomalie congenite, e nel 1963 ha presentato un documento: “Malformazioni non cardiache associate nei bambini con cardiopatia congenita”. Questo ha descritto 9 bambini che oltre alla cardiopatia congenita avevano caratteristiche facciali caratteristiche, deformità del torace e bassa statura.
Il Dott. John Opitz, un ex studente del Dott., Noonan, cominciò a chiamare la condizione “sindrome di Noonan” quando vide bambini che assomigliavano a quelli che il Dr. Noonan aveva descritto. Dr. Noonan ha prodotto un documento intitolato “Ipertelorismo con fenotipo Turner” nel 1968, dove ha studiato 19 pazienti che mostravano sintomi indicativi della sindrome di Noonan. Nel 1971 al Simposio dei difetti cardiovascolari, il nome “sindrome di Noonan” divenne ufficialmente riconosciuto.
















Lascia un commento