Introduzione
La disabilità intellettiva (ID) colpisce quasi l ‘ 1-2% della popolazione ed è il più comune disturbo dello sviluppo neurologico (NDD). Un numero sostanziale di pazienti con ID è risultato avere una causa genetica (rivisto in Bessa et al., 2012)., Le tecniche di analisi genome-wide attualmente utilizzate per lo studio dell’eziologia spesso portano all’identificazione di varianti quasi private molto rare, la raccolta di pazienti con alterazioni nello stesso gene è un aspetto cruciale della definizione di una nuova entità clinica.
All’inizio di quest’anno, i pazienti che ospitano mutazioni nel gene EBF3 sono stati descritti, presentando una sindrome dello sviluppo neurologico tra cui ID, atassia, ipotonia, lievi dismorfismi facciali e anomalie genitourinarie (OMIM 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., Il gene EBF3 (Early B cell Factor 3) codifica un membro della famiglia di fattori di trascrizione del fattore di cellule B precoce altamente conservato, espresso ad alti livelli nel sistema nervoso in via di sviluppo (dati recuperati dal portale GTEx). EBF3 è un bersaglio trascrizionale di ARX e dimostrato di essere regolato da NeuroD e ARX (Friocourt e Parnavelas, 2011). ARX codifica un fattore di trascrizione critico per lo sviluppo embrionale che, per molti anni, è stato associato a una vasta gamma di disturbi dello sviluppo neurologico., La compromissione intellettuale, il sistema nervoso centrale e le anomalie genito-urinarie osservate nei pazienti con entrambe le mutazioni in EBF3 e ARX potrebbero riflettere il contributo di entrambe le proteine agli stessi processi molecolari e cellulari (Chao et al., 2017). La funzione EBF3 è stata studiata anche in modelli animali. L’ablazione dei suoi ortologi nei vermi e nelle mosche porta a compromissione dello sviluppo neuronale (Prasad et al., 1998; Hattori et al., 2013)., Nei topi, l’eliminazione di Ebf3 porta a letalità neonatale e difetti di migrazione neuronale, con il fallimento del progetto dei neuroni olfattivi al bulbo olfattivo dorsale (Wang et al., 2004). Gli esatti meccanismi patogeni delle mutazioni EBF3 non sono ancora completamente chiariti, ma il tipo di varianti descritte finora suggeriscono che l’aploinsufficienza, il guadagno di funzione e il negativo dominante sono possibili meccanismi patogeni per le varianti descritte (Chao et al., 2017; Sleven et al., 2017).,
In questo lavoro contribuiamo con un paziente con la più piccola delezione (600 Kb) riportata fino ad oggi che interessa la totalità del gene EBF3 e con una presentazione clinica che si sovrappone a quella dei pazienti con varianti a singolo nucleotide EBF3 (SNVs). Inoltre, facciamo un confronto clinico dei pazienti con grandi eliminazioni terminali 10q precedentemente pubblicate e riportiamo che, nonostante le differenze di dimensioni, c’è una significativa sovrapposizione fenotipica tra i pazienti con queste alterazioni., Questi risultati si aggiungono alle attuali conoscenze sui disturbi correlati all’EBF3 e supportano l’aploinsufficienza dell’EBF3 come chiave nella sindrome dello sviluppo neurologico associata alle delezioni 10qter.
Materiali e metodi
Il paziente è stato accertato all’interno di un ampio studio sui disturbi dello sviluppo neurologico in Portogallo, in cui l’iscrizione dei pazienti e delle famiglie è stata effettuata dal medico di riferimento, le informazioni cliniche sono state raccolte in un database anonimo secondo l’Autorità portoghese per la protezione dei dati (CNPD) e, Il consenso informato per l’attuale paziente è stato fornito dalla madre per lo studio genetico e la pubblicazione dei risultati (comprese le foto). Lo studio è stato approvato dal comitato etico del Centro di Genetica Medica Dr. Jacinto Magalhães, National Health Institute Dr. Ricardo Jorge.
Il DNA genomico è stato estratto dal sangue periferico utilizzando Citogene® DNA isolation kit (Citomed, Portogallo). aCGH è stato eseguito utilizzando Agilent 180 K array (AMADID: 023363) contro un riferimento DNA diploide (MegaPoll Reference DNA di Kreatech, Kreatech Diagnostics, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., L’analisi è stata effettuata in una macchina PCR Real Time 7500-FAST (Applied Biosystems, Foster City, CA, USA) utilizzando Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) secondo le raccomandazioni del produttore e seguendo le linee guida generali per qPCR. La specificità di ciascuna reazione è stata verificata dalla generazione di una curva di fusione per ciascuno dei frammenti amplificati. L’efficienza del primer è stata calcolata mediante la generazione di una curva standard che corrisponde alla percentuale di efficienza normale accettata (primer utilizzati elencati nei dati supplementari)., I valori Ct ottenuti per ogni test sono stati analizzati nel software DataAssist™ (Applied Biosystems, Foster City, CA, USA).
Risultati
Qui descriviamo un paziente con una delezione de novo che colpisce EBF3. La paziente è una bambina di 11 anni con grave ID (global development quozient = 27 a 7 anni di età), nata da genitori non consanguinei e senza storia familiare di disturbi dello sviluppo neurologico. È nata dopo una gravidanza gemellare biamniotica bichorionica (la sorella è sana), con parto vaginale, a 35 settimane di gestazione. I parametri di nascita erano: peso, 1.830 g (P3); lunghezza, 42.,5 cm( P10); e OFC, 30,6 cm (P10), con un punteggio Apgar di 8/9 (1 ° e 5 ° min, rispettivamente). Il periodo neonatale era complicato con la sepsi e la diagnosi di sferocitosi ereditaria (ereditata da sua madre). Il ritardo dello sviluppo globale è stato notato nei primi mesi, con il controllo della testa raggiunto a 12 mesi, seduto a 18 mesi, camminata indipendente a 30 mesi e nessuna parola pronunciata all’età di 3 anni., Aveva pielonefrite a 19 mesi (ecografia renale non ha mostrato anomalie), reflusso gastroesofageo e otite media ricorrente, con perdita dell’udito conduttivo che ha richiesto un intervento chirurgico e un apparecchio acustico. L’epilessia è stata sospettata a 5 mesi (episodi di attività sospesa) ma l’EEG era normale.
È stata osservata per la prima volta all’età di 3 anni e 5 mesi (Figure 1A,B), momento in cui ha mostrato ipotonia muscolare, viso ipotonico, strabismo e ridotta sensibilità al dolore., Ha anche presentato lievi caratteristiche dismorfiche (Figura 1A): faccia triangolare, piccole orecchie basse con prominente anti-elica, sopracciglia arcuate, narici anteverted, punta nasale bulbosa, bocca piccola con angoli rivolti verso il basso, mento appuntito, collo corto, e cuscinetti fetali dito prominente, così come una bassa statura mite (89 cm, corrispondente a circa 2SD).

Figura 1. (A) Aspetto facciale del paziente a 3 anni e 5 mesi mostrando le orecchie piccole e basse con prominente anti-elica e (B) cuscinetti fetali nelle dita., (C) Aspetto facciale del paziente a 11 anni di età. (D) Evidenziati in grigio i 600 Kb cancellazione a 10q26.3 regione; uno zoom in della EBF3 gene nel DGV database rivela l’esistenza di 3 eliminazioni in 3 controlli che interessano i primi 6 esoni di EBF3 (NM_001005463); Cnv interno di questa regione, trovato in popolazioni di controllo includono le eliminazioni nvs825626 (presente in 1/31 individui), nvs552315 (presente in 1/17421 individui) e nsv552316 (presente in 1/31 individui). (E) La rappresentazione schematica delle trascrizioni EBF3.,
La risonanza magnetica cerebrale è stata eseguita a 6 anni, ma non sono state notate anomalie. All’età di 10 anni è stata rivalutata; aveva ancora otite media ricorrente, ma per il resto era in buona salute globale. La lingua era molto povera (due frasi di parole pronunciate dopo 8 anni). Ha avuto problemi di comportamento, con movimenti stereotipati (movimenti rotatori, masticazione di vestiti, retropulsione della testa), punteggio per grave disturbo dello spettro autistico (ADI-R e ADOS) a 7 anni; ha mostrato agitazione e comportamento aggressivo (auto ed etero) ed è stata medicata con farmaci antipsicotici., Un intervento chirurgico ortopedico è stato eseguito per pes planus. Le caratteristiche facciali erano simili a quelle descritte in precedenza, con incisivi centrali superiori distanziati (Figura 1C); aveva occhiali per strabismo e ipermetropia.
l’Analisi del DNA genomico mediante aCGH rivelato un de novo 600 kb cancellazione a 10p26.3 (Figura 1D) che interessano tre geni—MGMT (codifica l’enzima O-6-metilguanina-DNA metiltransferasi, coinvolti nella riparazione del DNA), EBF3 e GLRX (codifica glutaredoxin, un piccolo thioltransferase che rimuove le proteine GSH addotti), di cui EBF3 era più probabile associata a malattia del gene.,
Discussione
Il paziente presentato è stato analizzato per la prima volta da aCGH alcuni anni fa. Al momento dell’analisi aCGH, l’esistenza delle tre varianti presenti nel Database delle varianti genomiche (DGV)1 che interessano i primi cinque esoni del gene EBF3 (Figura 1E; Park et al., 2010; Cooper et al., 2011), così come l’assenza di altre malattie note che causano mutazioni in questo gene, ci portano a classificarlo come una variante di significato sconosciuto (VOUS). Tuttavia, le recenti pubblicazioni di mutazioni EBF3 (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) e le somiglianze cliniche con i casi riportati, ci portano a rivalutare la variante e ci fanno credere che la delezione di EBF3 possa in effetti rappresentare la malattia nel paziente. Uno degli aspetti che ha sollevato dubbi sulla patogenicità di questa variante in primo luogo è stata l’esistenza di controlli di popolazione con eliminazioni dei primi sei esoni di questo gene, in eterozigosi (dati recuperati dal database DGV a febbraio 2017) (Figura 1D). Anche se una trascrizione di EBF3 che inizia in Exon13b è elencata nel database Ensembl (EN00000440978.,1) (Figura 1E), che potrebbe spiegare come la cancellazione dei primi esoni potrebbe alla fine portare a un fenotipo normale, questa trascrizione esclude il dominio di legame del DNA di EBF3 e il suo modello di espressione e la sua rilevanza funzionale non sono stati caratterizzati. Al momento della rivalutazione, tuttavia, i CNV descritti da Cooper e colleghi (nsv552315 e nsv552316) (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) sono stati considerati alla soglia di rilevamento da SNP microarray e non possono essere la base per l’esclusione di un gene candidato, in particolare alla luce della forte evidenza genetica e funzionale per la rilevanza delle mutazioni EBF3 alla malattia (Evan Eichler, Greg Cooper e Bradley Coe, comunicazione personale).,
Il nostro paziente mostra molte somiglianze cliniche con pazienti precedentemente descritti con mutazioni in EBF3 (21 casi riassunti nella Tabella 1), come ritardo dello sviluppo globale, ritardo del linguaggio espressivo, ipotonia, aumento della soglia del dolore, problemi comportamentali e caratteristiche facciali caratteristiche (faccia lunga/triangolare, fronte grande, faccia ipotonica)., Tuttavia, anche se il nostro paziente ha avuto un ritardo significativo nello sviluppo motorio, non è stata rilevata alcuna atassia significativa (con alcune limitazioni nell’esame clinico, poiché il bambino non era cooperativo) e non erano presenti anomalie cerebellari nella risonanza magnetica cerebrale.

Tabella 1. Confronto clinico del caso in esame con i casi riportati con mutazioni puntiformi / indel nel gene EBF3.
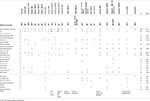
Tabella 2., Confronto clinico del caso in esame con i casi riportati con delezioni che interessano il cytoband 10q26.
Le varianti EBF3 descritte in letteratura all’inizio del 2017 includono mutazioni puntiformi predette come deleterie e piccole inserzioni e delezioni che portano alla delezione in frame di aminoacidi chiave o ad un frameshift, prevedibilmente causando il troncamento precoce della proteina risultante o il decadimento mediato da nonsense., Le mutazioni sono state concentrate su parti del gene che codifica per il dominio di legame al DNA di EBF3 e sono state previste attraverso diversi metodi per portare a una perdita di funzione di questo fattore di trascrizione, suggerendo così una ridotta funzione e aploinsufficienza come meccanismo alla base del disturbo dello sviluppo neurologico in questi pazienti. I topi knock-out per Ebf3 sono descritti per presentare letalità neonatale e difetti di migrazione neuronale, con il fallimento dei neuroni olfattivi di proiettare al bulbo olfattivo dorsale (Wang et al.,, 2004), ma nessuna descrizione è fatta di un fenotipo negli animali eterozigoti, che sono effettivamente presentati come controlli in molti degli esperimenti, quindi non supportano il modello di aploinsufficienza. Abbiamo fatto sforzi per ottenere e studiare il fenotipo neurosviluppo di questi animali, ma non hanno avuto successo, poiché la linea di knock-out Ebf3(O/E2) potrebbe essere stata interrotta (Joseph W. Lewcock, personal communication). Tuttavia, il caso attuale insieme ai pazienti riassunti nella Tabella 2, supportano l’ipotesi di aploinsufficienza di EBF3 come causa di malattia.,
In sintesi, la descrizione attuale rafforza la perdita di funzione / aploinsufficienza di EBF3 come causa della malattia dello sviluppo neurologico e rafforza l’associazione di questo gene con una sindrome clinica caratteristica all’interno di questo spettro.
Contributi dell’autore
FL ha eseguito gli studi molecolari e analizzato i dati molecolari. MG e GS hanno raccolto dati clinici. FL, JP e PM hanno esaminato tutti i casi di mutazione EBF3 in letteratura. FL, GS, JP e PM hanno redatto il documento. PM ha ottenuto finanziamenti per questo studio. Lo studio è stato eseguito sotto la direzione di PM.,
Finanziamento
FCT—Fundação para a Ciência e a Tecnologia nell’ambito dei progetti e delle borse di studio (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167 / 2012). Questo articolo è stato sviluppato nell’ambito del progetto NORTE-01-0145-FEDER-000013, sostenuto dal Programma operativo regionale del Portogallo settentrionale (NORTE 2020), nell’ambito dell’Accordo di partenariato Portogallo 2020, attraverso il Fondo europeo di sviluppo regionale (FEDER).
Dichiarazione di conflitto di interessi
JP è stato impiegato dalla società CGC Genetics.,
Gli altri autori dichiarano che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Ringraziamenti
Ringraziamo la paziente e la sua famiglia per la partecipazione agli studi genetici e per aver permesso questa pubblicazione. Siamo grati alla Dott. ssa Ana Maria Fortuna e alla Dott. ssa Margarida Reis Lima per aver reso possibile questo progetto e facilitando la nostra collaborazione con CGM.
Note a piè di pagina
1., ^Database di varianti genomiche Disponibili all’indirizzo: http://dgv.tcag.ca/dgv/app/home.
Friocourt, G., e Parnavelas, J. (2011). L’identificazione degli obiettivi Arx svela nuovi candidati per il controllo della migrazione e della differenziazione degli interneuroni corticali. Anteriore. Cellula. Neurosci. 5:28. doi: 10.3389 / fncel.2011.00028
PubMed Abstract / CrossRef Full Text / Google Scholar
















Lascia un commento