Bevezető
Szellemi fogyatékosság (ID) érint szinte 1-2% – át a lakosság, valamint a leggyakoribb fejlődési rendellenesség (NDD). Jelentős számú ID betegek azt találták, hogy a genetikai oka (felül Tessa et al., 2012)., Az etiológia vizsgálatára jelenleg alkalmazott Genom-szintű elemzési technikák gyakran nagyon ritka, szinte magánváltozatok azonosításához vezetnek, az ugyanazon génben bekövetkező változásokkal rendelkező betegek gyűjteménye az új klinikai egység meghatározásának kulcsfontosságú szempontja.
idén a betegek menedéket mutációk EBF3 gént írtak le, a bemutató egy fejlődési szindróma beleértve ID, ataxia, hypotonia, enyhe arc dysmorphisms, valamint urogenitális rendellenességek (OMIM 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., Az EBF3 (Early B cell Factor 3) gén az erősen konzervált korai B-cell faktor transzkripciós faktor család egy tagját kódolja, amelyet a fejlődő idegrendszerben magas szinten fejeznek ki (a GTEx Portalból származó adatok). Az EBF3 az ARX transzkripciós célpontja, amelyet a NeuroD és az ARX szabályoz (Friocourt and Parnavelas, 2011). Az ARX olyan transzkripciós tényezőt kódol, amely kritikus az embrionális fejlődéshez, amely évek óta számos neurodevelopmentális rendellenességhez kapcsolódik., Az ebf3 és ARX mutációkban egyaránt szenvedő betegeknél megfigyelt értelmi fogyatékosság, központi idegrendszer és genitourinary anomáliák tükrözhetik mindkét fehérje azonos molekuláris és sejtes folyamatokhoz való hozzájárulását (Chao et al., 2017). Az EBF3 funkciót állatmodellekben is tanulmányozták. A férgek és legyek ortológjainak ablációja a neuronális fejlődés károsodásához vezet (Prasad et al., 1998; Hattori et al., 2013)., Egerekben az Ebf3 kiütése újszülöttkori letalitáshoz és neuronális vándorlási hibákhoz vezet, a szaglási neuronok meghibásodása pedig a hátsó szaglóhagymához vezet (Wang et al., 2004). Az EBF3 mutációk pontos patogén mechanizmusai még nem teljesen tisztázottak, de az eddig leírt változatok típusa arra utal, hogy a haploinsufficiency, a funkció nyeresége és a domináns negatív lehetséges patogén mechanizmusok a leírt változatokhoz (Chao et al., 2017; Sleven et al., 2017).,
ebben A munkában részt vegyünk egy beteg a legkisebb törlés (600 Kb) arról számolt be, hogy dátuma érintő teljes EBF3 gén egy klinikai megjelenése, egymást átfedő, hogy a betegek EBF3 egyetlen nukleotid változatok (SNVs). Ezenkívül klinikai összehasonlítást végzünk a betegekkel a korábban közzétett nagy 10Q terminális törlésekkel, és beszámolunk arról, hogy a méretbeli különbségek ellenére jelentős fenotípusos átfedés van a betegek között ezekkel a változásokkal., Ezek az eredmények kiegészítik az EBF3-mal kapcsolatos rendellenességek jelenlegi ismereteit, és támogatják az EBF3 haploinsufficiency-t, mint a 10qter törléssel járó neurodevelopmentális szindróma kulcsát.
Anyagok, Módszerek
A beteg volt megállapítható belül egy nagy tanulmány fejlődési zavarok Portugáliában, ahol a beiratkozási a beteg, a család volt az a beküldő orvos, klinikai információkat gyűjtöttek a névtelen adatbázis szerint a portugál adatvédelmi Hatóság (CNPD), valamint írásbeli beleegyező volt, kapott minden résztvevő számára., A jelenlegi beteg beleegyezését adta az anya a genetikai vizsgálathoz és az eredmények közzétételéhez (beleértve a fényképeket is). A tanulmányt Dr. Jacinto Magalhães, a Dr. Ricardo Jorge Nemzeti Egészségügyi Intézet etikai bizottsága hagyta jóvá.
a genomikus DNS-t perifériás vérből extrahálták Citogene® DNS izolációs készlet (Citomed, Portugália) segítségével. az aCGH-t Agilent 180 k tömb (AMADID:023363) segítségével végezték diploid DNS referencia (Kreatech ‘ s MegaPoll Reference DNA, Kreatech Diagnostics, Amsterdam) ellen., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., Az elemzést egy 7500-as gyors valós idejű PCR gépen (Applied Biosystems, Foster City, CA, USA) hajtottuk végre a Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) segítségével a gyártó ajánlásai szerint, a qPCR általános irányelveinek megfelelően. Az egyes reakciók specifikusságát az egyes felerősített fragmensek olvadási görbéjének generálása igazolta. Az alapozó hatékonyságát az elfogadott normál hatékonysági százalékhoz illeszkedő szabványos görbe létrehozásával számítottuk ki (a kiegészítő adatokban felsorolt alapozók)., Az egyes tesztekhez kapott Ct értékeket a DataAssist™ szoftverben (Applied Biosystems, Foster City, CA, USA) elemeztük.
eredmények
itt leírjuk az EBF3-at befolyásoló de novo delécióval rendelkező beteget. A beteg egy 11 éves lány, súlyos ID-vel (globális fejlesztési hányados = 27 7 éves korban), nem véres szülőktől született, akiknek nincs családi anamnézisében neurodevelopmentális rendellenességek. Biamniotikus bichorionos iker terhesség után született (nővére egészséges), hüvelyi szülés útján, a terhesség 35 hetében. Születési paraméterei: súly, 1830 g (P3); Hossz, 42.,5 cm( P10); és OFC, 30,6 cm (P10), 8/9-es Apgar-pontszámmal (1. és 5. perc). Az újszülöttkori időszakot komplikálta a szepszis és az örökletes szferocitózis (az anyjától örökölt) diagnózisa. Globális fejlődési késleltetést figyeltek meg az első hónapokban, a fejkontroll 12 hónap, 18 hónap, 30 hónap független gyaloglás, 3 éves korban nem beszélt szavak., 19 hónapos korában pyelonephritis (vese ultrahang nem mutatott rendellenességet), gastrooesophagealis reflux és visszatérő otitis media, vezetőképes halláskárosodással, amely sebészeti beavatkozást és hallókészüléket igényelt. Az epilepsziát 5 hónapon belül gyanították (felfüggesztett aktivitási epizódok), de az EEG normális volt.
először 3 éves korában figyelték meg 5 hónap (1A, B ábra), ekkor izomhipotoniát, hipotóniás arcot, strabismust és csökkent fájdalomérzékenységet mutatott., Ő is bemutatott enyhe nála ezt a hibát funkciók (1A Ábra): háromszög alakú arc, kis alacsony-állítsa be a fülét a kiemelkedő anti-helix, ívelt szemöldök, anteverted orrlyukam, hagymás orr tipp, kis száját lefelé fordult sarkok, hegyes álla, rövid nyak, prominens ujját magzati párna, valamint egy enyhe alacsony termetű (89 cm, megfelelő körül 2SD).

1.ábra. A) A beteg arckifejezése 3 éves és 5 hónapos korban, a kicsi és alacsony fülű füleket mutatva, kiemelkedő anti-helix-szel és (B) magzati párnákkal az ujjakban., C) a beteg arckifejezése 11 éves korban. (D) szürke színnel kiemelve a 600 Kb-os Törlés a 10q26.3 régióban; az EBF3 gén nagyítása a DGV adatbázisban 3 törlés létezését tárja fel 3 kontrollban, amelyek befolyásolják az EBF3 első 6 exonját (NM_001005463); a kontrollpopulációkban található CNV-k közé tartoznak az nvs825626 törlések (jelen 1/31 egyénben), nvs552315 (jelen 1/17421 egyénben) és nsv552316 (jelen 1/31 egyének). E) az EBF3 átiratok sematikus ábrázolása.,
az agyi MRI-t 6 év alatt végezték el, de nem észleltek rendellenességeket. 10 éves korában újraértékelték; még mindig visszatérő otitis media volt, de egyébként jó globális egészségi állapotban volt. A nyelv nagyon gyenge volt (két szó mondat 8 év után). Viselkedési problémái voltak, sztereotip mozgásokkal (forgó mozgások, ruhák rágása, fejrepropulzió), súlyos autizmus spektrum zavar (ADI-R és ADOS) pontozása 7 év alatt; agitációt és agresszív viselkedést mutatott (auto és hetero), és antipszichotikus gyógyszerekkel kezelték., Ortopédiai műtétet végeztek a pes planus számára. Az arcvonások hasonlóak voltak a korábban leírtakhoz, a felső középső metszőfogak (1C ábra) egymástól távol helyezkednek el; a strabismus és a hypermetropia szemüvegei voltak.
az aCGH genomikus DNS analízise 10p26.3 (1D ábra) esetén de novo 600 kb deléciót mutatott ki, amely három gént érint—Mgmt (az O-6-metilguanin-DNS metiltranszferáz kódolása, amely részt vesz a DNS javításában), EBF3 és GLRX (a glutaredoxin kódolása, egy kis tioltranszferáz, amely eltávolítja a GSH adductokat), amelyek közül az EBF3 volt a legvalószínűbb betegséggel összefüggő gén.,
Vita
a bemutatott beteget először az aCGH elemezte néhány évvel ezelőtt. Az aCGH analízis idején az EBF3 gén első öt exonját (1e ábra; Park et al., 2010; Cooper et al., 2011), valamint más ismert betegség hiánya, amely mutációkat okoz ebben a génben, arra készteti bennünket, hogy ismeretlen jelentőségű változatnak (VOUS) minősítsük. Azonban a legutóbbi publikációk EBF3 mutációk (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) és a klinikai hasonlóságok a jelentett esetekkel, arra késztetnek bennünket, hogy újra értékeljük a változatot, és elhitessük velünk, hogy az EBF3 deléció valójában a beteg beteg betegségének elszámolása lehet. Az egyik szempont, amely kétségeket vetett fel e változat patogenitásával kapcsolatban, elsősorban a gén első hat exonjának törlését hordozó populációs kontrollok létezése volt heterozigozitásban (a DGV adatbázisból 2017 februárjától származó adatok) (1D ábra). Annak ellenére, hogy az Exon13b-ben kezdődő EBF3 átirata szerepel az Ensembl adatbázisban (ENST0000440978.,1) (1e ábra), amely megmagyarázhatja, hogy az első exonok deléciója végül normális fenotípushoz vezethet, ez az átirat kizárja az EBF3 DNS-kötő doménjét, expressziós mintáját és funkcionális relevanciáját nem jellemezték. Az újraértékelés során azonban a Cooper és kollégái (nsv552315 és nsv552316) által leírt CNV-K (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) az SNP microarray kimutatási küszöbén állt, és nem lehet a jelölt gén kizárásának alapja, különös tekintettel az EBF3 mutációk betegségekre való relevanciájának erős genetikai és funkcionális bizonyítékaira (Evan Eichler, Greg Cooper és Bradley Coe, személyes kommunikáció).,
A beteg azt mutatja, sok klinikai hasonlóságot mutat a korábban leírt betegek mutációk EBF3 (21 esetben Táblázat foglalja össze 1), mint például a globális fejlődési késés, késleltetett kifejező beszéd, hypotonia, fájdalom küszöb, viselkedési problémák, karakteres arcvonások (hosszú/háromszög alakú arc, nagy homloka, hipotóniás arc)., Annak ellenére azonban, hogy páciensünknek jelentős késése volt a motoros fejlődésben, nem észleltek jelentős ataxiát (a klinikai vizsgálat bizonyos korlátozásaival, mivel a gyermek nem volt együttműködő), és az agyi MRI-ben nem voltak cerebelláris anomáliák.

1.táblázat. A jelen eset klinikai összehasonlítása a jelentett esetekkel az EBF3 gén pontmutációival/indeljeivel.
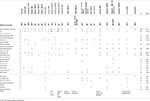
2.táblázat., A jelen eset klinikai összehasonlítása a jelentett esetekkel a 10q26 cytobandot érintő törlésekkel.
A EBF3 változatok leírása az irodalomban az elején 2017 tartalmazza pontmutációk megjósolta, hogy káros, illetve apró betoldások, illetve törléséhez, ami a keret törlése gombot úton, vagy az olvasási kiszámíthatóan okoz korai vágásos a keletkező fehérje vagy ostobaság-mediált bomlás., A mutációk az EBF3 DNS-kötő doménjét kódoló gén egyes részeire összpontosultak, és különböző módszerekkel megjósolták, hogy ennek a transzkripciós faktornak a funkcióvesztéséhez vezetnek, ami arra utal, hogy csökkent a funkció és a haploinsufficiencia, mint a neurodevelopmentális zavar alapjául szolgáló mechanizmus ezekben a betegekben. Knock-out egerek Ebf3 ismertetett, hogy jelen újszülött halálos vagy neuronális migráció hibák, a kudarc, a szagló neuronok, hogy a projekt a háti szagló izzó (Wang et al.,, 2004), de nincs leírás a heterozigóta állatok fenotípusáról, amelyeket valójában számos kísérletben kontrollként mutatnak be, így nem támogatják a haploinsufficiency modellt. Igyekeztünk ezeknek az állatoknak a neurodevelopmentális fenotípusát megszerezni és tanulmányozni, de nem voltunk sikeresek, mivel az Ebf3(O/E2) knock-out vonal megszakadhatott (Joseph W. Lewcock, személyes kommunikáció). A jelenlegi ESET azonban a 2. táblázatban összefoglalt betegekkel együtt alátámasztja az EBF3 haploinsufficiencia hipotézisét, mint betegséget okozó betegséget.,
összefoglalva, a jelenlegi leírás megerősíti az EBF3 funkcióvesztést / haploinsufficienciát, mint neurodevelopmentális betegség okát, és megerősíti ennek a génnek a társulását egy jellegzetes klinikai szindrómával ezen a spektrumon belül.
szerzői hozzájárulások
FL elvégezte a molekuláris vizsgálatokat és elemezte a molekuláris adatokat. Az MG és a GS klinikai adatokat gyűjtött. Az FL, A JP és a PM áttekintette az összes EBF3 mutációs esetet a szakirodalomban. FL, GS, JP és PM szerkesztette a lapot. A PM finanszírozást kapott ehhez a tanulmányhoz. A vizsgálatot PM irányítása alatt végezték.,
finanszírozás
FCT—Fundação para a Ciência e a Tecnologia a projekteken és ösztöndíjakon belül (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Ezt a cikket a NORTE projekt keretében fejlesztették ki-01-0145-FEDER-000013, amelyet az Észak-portugáliai Regionális Operatív Program (NORTE 2020) támogat a Portugália 2020 partnerségi megállapodás keretében, az Európai Regionális Fejlesztési Alap (FEDER) révén.
összeférhetetlenségi nyilatkozat
a JP-t a CGC Genetics cég alkalmazta.,
a többi szerző kijelenti, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
elismerések
szeretnénk köszönetet mondani a betegnek és családjának a genetikai vizsgálatokban való részvételért és a kiadvány engedélyezéséért. Hálásak vagyunk Dr. Ana Maria Fortuna-nak és Dr. Margarida Reis Lima-nak, hogy lehetővé tették ezt a projektet, és megkönnyítettük a CGM-mel való együttműködésünket.
1., A genomikai változatok adatbázisa a következő címen érhető el: http://dgv.tcag.ca/dgv/app/home.
Friocourt, G., and Parnavelas, J. (2011). Az Arx-célok azonosítása új jelölteket mutat be az agykérgi belső migráció és a differenciálás ellenőrzésére. Elöl. Cell. Neurológus. 5:28. doi: 10.3389 / fncel.2011.00028
PubMed Abstract / CrossRef teljes szöveg / Google Scholar
















Vélemény, hozzászólás?