Introduction
la déficience intellectuelle (ID) affecte près de 1 à 2% de la population et est le trouble neurodéveloppemental (NDD) le plus courant. Un nombre important de patients atteints D’ID ont une cause génétique (examiné dans Bessa et al., 2012)., Les techniques d’analyse à l’échelle du génome actuellement utilisées pour l’étude de l’étiologie conduisent souvent à l’identification de très rares variants presque privés, la collecte de patients présentant des altérations du même gène étant un aspect crucial de la définition d’une nouvelle entité clinique.
plus tôt cette année, des patients porteurs de mutations dans le gène EBF3 ont été décrits, présentant un syndrome neurodéveloppemental comprenant ID, ataxie, hypotonie, dysmorphismes faciaux légers et anomalies génito-urinaires (OMIM 617330) (Chao et al., 2017; Harms et coll. En 2017; Sleven et coll., 2017)., Le gène EBF3 (Early B cell Factor 3) code un membre de la famille des facteurs de transcription du facteur de cellules B précoces hautement conservés, exprimé à des niveaux élevés dans le système nerveux en développement (données extraites du portail GTEx). EBF3 est une cible transcriptionnelle D’ARX, et il a été démontré qu’elle est régulée par NeuroD et ARX (Friocourt et Parnavelas, 2011). ARX code un facteur de transcription critique pour le développement embryonnaire qui, depuis de nombreuses années, a été associé à un large éventail de troubles neurodéveloppementaux., La déficience intellectuelle, le système nerveux central et les anomalies génito-urinaires observés chez les patients présentant les deux mutations dans EBF3 et ARX pourraient refléter la contribution des deux protéines aux mêmes processus moléculaires et cellulaires (Chao et al., 2017). La fonction EBF3 a également été étudiée dans des modèles animaux. L’Ablation de ses orthologues chez les vers et les mouches entraîne une altération du développement neuronal (Prasad et al., 1998; Hattori et coll., 2013)., Chez la souris, l’élimination D’Ebf3 entraîne une létalité néonatale et des défauts de migration neuronale, avec une défaillance du projet des neurones olfactifs vers le bulbe olfactif dorsal (Wang et al., 2004). Les mécanismes pathogènes exacts des mutations EBF3 ne sont pas encore complètement élucidés, mais le type de variants décrits jusqu’à présent suggèrent que l’haploinsuffisance, le gain de fonction et le négatif dominant sont des mécanismes pathogènes possibles pour les variants décrits (Chao et al. En 2017; Sleven et coll., 2017).,
dans ce travail, nous contribuons avec un patient avec la plus petite délétion (600 Kb) rapportée à ce jour affectant la totalité du gène EBF3 et avec une présentation clinique chevauchant celle des patients avec des variants nucléotidiques simples EBF3 (SNVs). De plus, nous effectuons une comparaison clinique des patients avec de grandes suppressions terminales de 10Q publiées précédemment et signalons que, malgré les différences de taille, il existe un chevauchement phénotypique significatif entre les patients présentant ces altérations., Ces résultats ajoutent aux connaissances actuelles sur les troubles liés à EBF3 et soutiennent L’haploinsuffisance EBF3 comme clé dans le syndrome neurodéveloppemental associé aux délétions de 10QTER.
matériaux et méthodes
le patient a été vérifié dans le cadre d’une vaste étude sur les troubles neurodéveloppementaux au Portugal, dans laquelle l’inscription des patients et des familles a été effectuée par le médecin référent, des informations cliniques ont été recueillies dans une base de données anonyme selon L’Autorité portugaise de Protection des données (CNPD) et un, Le consentement éclairé du patient actuel a été fourni par la mère pour l’étude génétique et la publication des résultats (y compris les photos). L’étude a été approuvée par le Comité d’éthique du Centre de Génétique médicale Dr Jacinto Magalhães, Institut national de la santé Dr Ricardo Jorge.
L’ADN génomique a été extrait du sang périphérique à l’aide du kit D’isolement D’ADN Citogene® (Citomed, Portugal). aCGH a été réalisée en utilisant Agilent 180 K array (AMADID: 023363) contre une référence d’ADN diploïde (Kreatech’s MegaPoll Reference DNA, Kreatech Diagnostics, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., L’analyse a été réalisée sur une machine de PCR en temps réel 7500-FAST (Applied Biosystems, Foster City, CA, USA) utilisant Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) selon les recommandations du fabricant et suivant les directives générales pour qPCR. La spécificité de chaque réaction a été vérifiée par la génération d’une courbe de fusion pour chacun des fragments amplifiés. L’efficacité des amorces a été calculée par la génération d’une courbe standard correspondant au pourcentage d’efficacité normale accepté (amorces utilisées énumérées dans les données supplémentaires)., Les valeurs Ct obtenues pour chaque test ont été analysées dans le logiciel DataAssist™ (Applied Biosystems, Foster City, CA, USA).
résultats
Nous décrivons ici un patient avec une délétion de novo affectant EBF3. La patiente est une fille de 11 ans avec un ID sévère (quotient de développement global = 27 à 7 ans), née de parents non consanguins et sans antécédents familiaux de troubles neurodéveloppementaux. Elle est née après une grossesse gémellaire biamniotique bichorionique (sa sœur étant en bonne santé), par accouchement vaginal, à 35 semaines de gestation. Les paramètres de naissance étaient: poids, 1,830 g (P3); Longueur, 42.,5 cm (P10); et OFC, 30,6 cm (P10), avec un score Apgar de 8/9 (1ère et 5ème min, respectivement). La période néonatale était compliquée par la septicémie et le diagnostic de sphérocytose héréditaire (héritée de sa mère). Un retard global de développement a été noté dans les premiers mois, avec un contrôle de la tête atteint à 12 mois, assis à 18 mois, marche indépendante à 30 mois et aucun mot prononcé à l’âge de 3 ans., Elle a eu une pyélonéphrite à 19 mois (l’échographie rénale n’a montré aucune anomalie), un reflux gastro-oesophagien et une otite moyenne récurrente, avec une perte auditive conductrice nécessitant une intervention chirurgicale et une aide auditive. L’épilepsie a été suspectée à 5 mois (épisodes d’activité suspendue) mais l’EEG était normale.
elle a été observée pour la première fois à l’âge de 3 ans et 5 mois (Figures 1A,B), période à laquelle elle a présenté une hypotonie musculaire, un visage hypotonique, un strabisme et une sensibilité réduite à la douleur., Elle a également présenté des caractéristiques dysmorphiques légères (Figure 1A): visage triangulaire, petites oreilles basses avec anti-hélice proéminente, sourcils arqués, narines antéverses, bout nasal bulbeux, petite bouche avec coins rabattus, menton pointu, cou court et coussinets fœtaux proéminents, ainsi qu’une petite taille (89 cm, correspondant à environ 2SD).

la Figure 1. (A) l’apparence du visage du patient à 3 ans et 5 mois montrant les oreilles petites et basses avec anti-hélice proéminente et (B) coussinets fœtaux dans les doigts., (C) l’apparence du visage du patient à l’âge de 11 ans. (D) a mis en évidence en gris la délétion de 600 Kb dans la région 10q26.3; un zoom avant du gène EBF3 dans la base de données DGV révèle l’existence de 3 délétions chez 3 témoins qui affectent les 6 premiers exons D’EBF3 (NM_001005463); les CNV dans cette région trouvés dans les populations témoins comprennent les délétions nvs825626 (présente chez 1/31 individus), nvs552315 (présente chez 1/17421 individus) et nsv552316 (présent chez 1/31 individus). E) la représentation schématique des transcriptions EBF3.,
l’IRM Cérébrale a été réalisée à 6 ans, mais aucune anomalie n’a été notée. À l’âge de 10 ans, elle a été réévaluée; elle avait encore une otite moyenne récurrente, mais était en bonne santé mondiale. La langue était très pauvre (deux phrases prononcées après 8 ans). Elle avait des problèmes de comportement, avec des mouvements stéréotypés (mouvements rotatifs, mastication des vêtements, rétropulsion de la tête), marquant pour le trouble du spectre autistique sévère (ADI-R et ADO) à 7 ans; elle affichait une agitation et un comportement agressif (auto et hétéro) et était médicamentée avec des antipsychotiques., Une chirurgie orthopédique a été réalisée pour pes planus. Les traits du visage étaient similaires à ceux décrits précédemment, avec des incisives centrales supérieures espacées (Figure 1C); elle avait des lunettes pour le strabisme et l’hypermétropie.
L’analyse de L’ADN génomique par l’aCGH a révélé une délétion de novo de 600 kb à 10p26.3 (Figure 1d) affectant trois gènes—MGMT (codant pour L’enzyme O-6-méthylguanine-ADN méthyltransférase, impliquée dans la réparation de l’ADN), EBF3 et GLRX (codant pour la glutarédoxine, une petite thioltransférase qui élimine les adduits de la protéine GSH), ,
Discussion
le patient présenté a été analysé pour la première fois par l’aCGH il y a quelques années. Au moment de l’analyse aCGH, l’existence des trois variants présents dans la base de données des Variants génomiques (DGV)1 affectant les cinq premiers exons du gène EBF3 (Figure 1e; Park et al., 2010; Cooper et coll., 2011), ainsi que l’absence d’autres maladies causant des mutations dans ce gène, nous conduire à le classer comme une variante de signification inconnue (VOUS). Cependant, les publications récentes de mutations EBF3 (Chao et al., 2017; Harms et coll. En 2017; Sleven et coll.,, 2017) et les similitudes cliniques avec les cas rapportés, nous conduisent à réévaluer la variante et nous font croire que la suppression de L’EBF3 peut en fait expliquer la maladie chez le patient. L’un des aspects qui ont soulevé des doutes sur la pathogénicité de cette variante en premier lieu était l’existence de contrôles de population portant des délétions des six premiers exons de ce gène, en hétérozygotie (données extraites de la base de données DGV en février 2017) (Figure 1D). Même si une transcription D’EBF3 commençant par Exon13b est répertoriée dans la base de données Ensembl (Enst00000440978.,1) (Figure 1E), ce qui pourrait expliquer comment la délétion des premiers exons pourrait éventuellement aboutir à un phénotype normal, cette transcription exclut le domaine de liaison à L’ADN D’EBF3, et son modèle d’expression et sa pertinence fonctionnelle n’ont pas été caractérisés. Cependant, après réévaluation, les VNC décrites par Cooper et ses collègues (nsv552315 et nsv552316) (Chao et al., 2017; Harms et coll. En 2017; Sleven et coll.,, 2017) ont été considérés comme étant au seuil de détection par les puces SNP et ne peuvent pas être la base de l’exclusion d’un gène candidat, en particulier à la lumière des preuves génétiques et fonctionnelles solides de la pertinence des mutations EBF3 pour la maladie (Evan Eichler, Greg Cooper et Bradley Coe, communication personnelle).,
notre patient présente de nombreuses similitudes cliniques avec les patients précédemment décrits avec des mutations dans EBF3 (21 cas résumés dans le tableau 1), tels que retard de développement global, retard de la parole expressive, hypotonie, augmentation du seuil de douleur, problèmes de comportement et traits faciaux caractéristiques (visage long / triangulaire, grand front, visage hypotonique)., Cependant, même si notre patient avait un retard important dans le développement moteur, aucune ataxie significative n’a été détectée (avec certaines limitations dans l’examen clinique, car l’enfant n’était pas coopératif), et aucune anomalie cérébelleuse n’était présente dans L’IRM cérébrale.

le Tableau 1. Comparaison clinique du cas présent avec les cas rapportés avec des mutations ponctuelles/indels dans le gène EBF3.
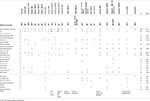
le Tableau 2., Comparaison clinique du cas présent avec les cas signalés de délétions affectant le cytobande 10q26.
les variantes EBF3 décrites dans la littérature au début de 2017 comprennent des mutations ponctuelles prédites comme étant délétères et de petites insertions et délétions conduisant à la suppression dans le cadre d’aminoacides clés ou à un frameshift, provoquant de manière prévisible une troncature précoce de la protéine résultante ou une désintégration Non-sensée., Les mutations ont été concentrées sur des parties du gène codant pour le domaine de liaison à L’ADN D’EBF3, et ont été prédites par différentes méthodes pour conduire à une perte de fonction de ce facteur de transcription, suggérant ainsi une fonction réduite et une haploinsuffisance comme mécanisme sous-jacent à la perturbation neurodéveloppementale chez ces patients. Les souris Knock-out pour Ebf3 sont décrites pour présenter une létalité néonatale et des défauts de migration neuronale, avec une incapacité des neurones olfactifs à se projeter vers le bulbe olfactif dorsal (Wang et al.,, 2004), mais aucune description n’est faite d’un phénotype chez les animaux hétérozygotes, qui sont en fait présentés comme des témoins dans de nombreuses expériences, ne supportant donc pas le modèle d’haploinsuffisance. Nous avons fait des efforts pour obtenir et étudier le phénotype neurodéveloppemental de ces animaux, mais nous n’avons pas réussi, car la lignée Ebf3(O/E2) knock-out a peut-être été abandonnée (Joseph W. Lewcock, communication personnelle). Cependant, le cas actuel ainsi que les patients résumés dans le tableau 2, soutiennent l’hypothèse de L’haploinsuffisance EBF3 comme cause de maladie.,
En résumé, la description actuelle renforce la perte de fonction / haploinsuffisance EBF3 comme cause de maladie neurodéveloppementale, et renforce l’association de ce gène avec un syndrome clinique caractéristique dans ce spectre.
contributions des auteurs
FL a effectué les études moléculaires et analysé les données moléculaires. MG et GS ont recueilli des données cliniques. FL, JP et PM ont examiné tous les cas de mutation EBF3 dans la littérature. FL, GS, JP et PM ont rédigé le document. PM a obtenu des fonds pour cette étude. L’étude a été réalisée sous la direction de PM.,
Financement
FCT—Fundação para a Ciência e a Tecnologia dans le cadre des projets et bourses (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167 / 2012). Cet article a été développé dans le cadre du projet NORTE-01-0145-FEDER-000013, soutenu par le programme opérationnel régional du Nord du Portugal (NORTE 2020), dans le cadre de L’accord de partenariat Portugal 2020, par l’intermédiaire du Fonds européen de développement régional (FEDER).
déclaration de conflit d’intérêts
JP était employé par la société CGC Genetics.,
les autres auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière pouvant être interprétée comme un conflit d’intérêts potentiel.
Remerciements
Nous tenons à remercier la patiente et sa famille d’avoir participé aux études génétiques et d’avoir permis cette publication. Nous sommes reconnaissants au Dr Ana Maria Fortuna et au Dr Margarida Reis Lima d’avoir rendu ce projet possible et de faciliter notre collaboration avec CGM.
Notes de bas de page
1., ^De la base de données de Variantes Génomiques Disponible sur: http://dgv.tcag.ca/dgv/app/home .
Friocourt, G., et Parnavelas, J. (2011). L’Identification des cibles Arx dévoile de nouveaux candidats pour contrôler la migration et la différenciation des interneurones corticaux. Devant. Cellule. Neurosci. 5:28. doi: 10.3389 / fncel.2011.00028
PubMed résumé / CrossRef Texte intégral / Google Scholar
















Laisser un commentaire