Mužský Turnerův syndrom, Noonanové-Ehmke syndrom, Turner-like syndrom, Ullrich-Noonan syndrom,
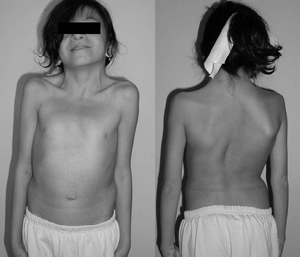
12-rok-stará dívka s Noonan syndromem. Typický webbed krk. Dvojitá strukturální křivka s deformitou žeber.,. bylo však potvrzeno, s genetické testování
Cardiofaciocutaneous syndrom, Turnerův syndrom, Costello syndrom, neurofibromatóza typu 1,
na Základě příznaků
Růstový hormon
Závisí na závažnosti srdeční problémy,
1 ze 100 (1 za 2000 závažné onemocnění)
syndrom Noonanové (NS) je genetická porucha, která může představovat s mírně neobvyklé rysy obličeje, krátké výšky, vrozená srdeční onemocnění, problémy s krvácením, a skeletální malformace., Funkce obličeje zahrnují široce rozložené oči, světlé oči, uši s nízkým nastavením, krátký krk a malou dolní čelist. Srdeční problémy mohou zahrnovat stenózu plicní chlopně. Prsní kost může buď vyčnívat nebo být potopena, zatímco páteř může být abnormálně zakřivená. Inteligence v syndromu je často normální. Komplikace NS mohou zahrnovat leukémii.
řada genetických mutací může mít za následek Noonanův syndrom. Tento stav může být zděděn od rodičů osoby jako autosomálně dominantní stav nebo se může objevit jako nová mutace., Noonanův syndrom je typ Rasopatie, jejíž základní mechanismus zahrnuje nadměrnou aktivitu v signální dráze buněk RAS / MAPK. Diagnóza může být podezřelá na základě příznaků, lékařského zobrazování a krevních testů. Potvrzení může být dosaženo genetickým testováním.
není znám žádný lék na NS. Léčba je založena na symptomech a základních problémech a může být vyžadována další podpora ve škole. Terapie růstovým hormonem během dětství může zvýšit konečnou výšku postižené osoby. Dlouhodobé výsledky obvykle závisí na závažnosti srdečních problémů.,
odhadem 1 z 1000 lidí je NS mírně postiženo, zatímco asi 1 z 2000 má závažnější formu stavu. Zdá se, že muži jsou postiženi častěji než ženy. Stav byl poprvé popsán v roce 1883 a byla pojmenována po Americké pediatrické kardiolog Jacqueline Noonan, který popsal další případy v roce 1963.
příznaky

Abnormální funkce Noonan syndromem ve věku 3 měsíců: Poznámka obočí šikmo a levé víčko klesá.,

Abnormální funkce Noonan syndromem ve věku 3 měsíců: Poznámka: nízko posazené, posteriorly otáčet, a abnormálně tvarované ucho.
nejčastějšími příznaky, které vedou k diagnóze noonanova syndromu, jsou jedinečné vlastnosti obličeje a muskuloskeletální rysy. Charakteristiky obličeje jsou nejvýraznější v dětství a u mnoha lidí s Noonanovým syndromem jsou s věkem méně patrné.,
Některé charakteristické rysy Noonan syndrom zahrnuje velkou hlavu s přebytečné kůže na zadní straně krku, nízké čelo, na šíji, vysoký vlasů v přední části hlavy, trojúhelníkový tvar obličeje, široké čelo a krátké, webbed krku.
v očích je hypertelorismus (široce nastavené oči) definující charakteristikou, přítomnou u 95% lidí s noonanovým syndromem., To může být doprovázeno epicanthal záhyby (extra kožní řasy u vnitřního koutku oka), ptóza (pokleslé oční víčka), proptosis (vypouklé oči), strabismus (dovnitř nebo ven otáčení očí), nystagmus (trhavé pohyby očí) a lomu vizuální chyby.
nos může být malý, široký a obrácený.
vývoj úst může být také ovlivněn noonanovým syndromem., To může mít za následek hluboce rýhované philtrum (horní ret linka) (přes 90%), mikrognacie (podměrečné dolní čelisti), vysoce klenuté patro, artikulační obtíže (zuby nejsou line up), což může vést k zubní problémy. Podobně jako u výše uvedených svalových projevů může být v ústech pozorována špatná kontrola jazyka.,
Skin
Kožní příznaky a symptomy v Noonan syndrom patří lymfedém (mízní otok končetin), keloidní formace, nadměrné tvorbě jizev, hyperkeratóza (příliš rozvinutá vnější vrstvy kůže), pigmentové névy (tmavě pigmentované kožní skvrny), a onemocnění pojivové tkáně,
Pohybového aparátu
Abnormality končetin a končetinách se mohou objevit v Noonan syndrom. To se může projevit jako bez obalu skončil prsty, extra polstrování na prstech rukou a nohou, otok v zadní části rukou a vrcholky nohy a lokte valgus (široký přenášení úhlu, lokty).,
pro krátkou postavu je růstový hormon někdy kombinován s IGF-1 (nebo jako alternativa, IGF-1 jako samostatný) lze použít k rychlejšímu dosažení zvýšené výšky/konečné výšky. Konečná výška dospělých jedinců s noonanovým syndromem je u mužů asi 161-167 cm a u žen 150-155 cm, což se blíží dolní hranici normálu.
abnormality páteře mohou být přítomny až 30% času, což může vyžadovat chirurgický zákrok k nápravě ve více než 60% těchto případů., Další muskuloskeletální projevy v Noonan syndrom jsou spojeny s nediferencované onemocnění pojivové tkáně poruchy, které mohou být spojeny s kloubní kontraktury (těsnost) nebo kloubní hypermobilita (uvolnění). Další faktory mohou být přítomny ve formě wingingu lopatky, skoliózy, prominence prsní kosti (pectus carinatum), deprese prsní kosti (pectus excavatum). Svalové abnormality mohou prezentovat jako hypotonie (snížený svalový tonus), což může vést k lordózy (zvýšená dutý vzadu) z důvodu špatné břišní svalový tonus.,
heart
Noonanův syndrom je druhou nejčastější syndromickou příčinou vrozených srdečních chorob. To zahrnuje plicní chlopenní stenózu (50-60%), defekty síňového septa (10-25%), defekty komorového septa (5-20%) a hypertrofickou kardiomyopatii (12-35%).
plíce
u některých lidí byla hlášena restriktivní funkce plic.
gastrointestinální
řada různých gastrointestinálních (GI) příznaků byla spojena s Noonanovým syndromem., Patří mezi ně potíže s polykáním, nízkou motilitu střev, gastroparéza (zpožděné vyprazdňování žaludku), střevní malrotace, nebo prudké a časté zvracení. Tyto zažívací problémy mohou vést ke snížení chuti k jídlu, selhání se daří od dětství do puberty (75%), a občas potřeba krmení trubice.{ To bylo spojeno s Von Willebrandova choroba, Amegakaryocytic trombocytopenie (nízký počet krevních destiček), prodloužený aktivovaný parciální tromboplastinový čas, kombinované vady koagulace., Pokud jsou přítomny, tyto Noonan-syndrom doprovodné poruchy mohou být spojeny s predispozicí k modřinám snadno, nebo krvácení.
neurologické
občas se může objevit Chiariho malformace (typ 1),což může vést k hydrocefalu. Byly také hlášeny záchvaty.
Příčiny

NS se obvykle dědí autosomálně dominantní vzor s variabilní exprese.,
Opakování v sourozenci a zdánlivý přenos z rodiče na dítě má dlouhé navrhl genetická porucha s autozomálně dominantní dědičnost a variabilní exprese. Je známo, že mutace v signálních drahách Ras/mitogen aktivované protein kinázy jsou zodpovědné za přibližně 70% případů NS.
osoby s NS mají až 50% šanci jej předat svým potomkům., Skutečnost, že postižený rodič není vždy určeny pro děti s NS navrhuje několik možností:
- Projevy by mohly být tak jemné, jako jít nepovšimnutý (variabilní expresivita)
- NS je heterogenní, zahrnující více než jednu podobný stav z odlišných příčin, a některé z nich nemusí být dědičná.
- vysoký podíl případů může představovat nové, sporadické mutace.,
| Typ | on-Line Mendelian Inheritance in Man databáze | Gen | Rok | Místo | % případů | Popis | Refs. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | PTPN11 gen kóduje protein tyrosin fosfatázy SHP-2., Tento protein je složkou několika intracelulárních signálních transdukcí drah zapojených v embryonální vývoj, které modulují buněčné dělení, diferenciace a migrace, včetně jedné zprostředkované receptor pro epidermální růstový faktor, což je důležité při tvorbě semilunární chlopně. duplikace chromozomové oblasti obsahující PTPN11 může také vést k NS., |
|
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
Heterozygotní mutace v NRAS, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, a CBI byly také spojeny s menší procento NS a souvisejících fenotypů.
stav známý jako „neurofibromatóza-Noonanův syndrom“ je spojen s neurofibrominem.
diagnóza
NS může být geneticky potvrzena přítomností kterékoli ze známých mutací uvedených výše., Nicméně, navzdory identifikaci 14 příčinných genů, absence známé mutace nevylučuje diagnózu, protože více, dosud neobjevených genů může způsobit NS. Diagnóza NS je tedy stále založena na klinických rysech. Jinými slovy, to je děláno, když lékař cítí, že člověk má dostatek funkcí, které zaručují štítek. Hlavní hodnoty genetické diagnostiky spočívají v tom, že řídí další lékařská a vývojová hodnocení, vylučuje další možná vysvětlení vlastností a umožňuje přesnější odhady rizik recidivy., S více genotyp-fenotyp srovnávací studie prováděny, pozitivní genetická diagnóza pomůže lékaři, aby si být vědomi možných anomálií specifických pro určité mutace genu. Například zvýšení hypertrofické kardiomyopatie je pozorováno u lidí s mutací KRAS a zvýšené riziko juvenilní myelomonocytární leukémie existuje pro mutaci PTPN11. V budoucnu mohou studie vést k cílené léčbě symptomů NS, které závisí na tom, jakou genetickou mutaci má člověk.,
Před narozením
Prenatální funkce, které by mohly vést lékaři považují diagnózu Noonan syndrom, patří cystická hygrom, zvýšená nuchální translucence, pleurální výpotek a edém.
Diferenciální diagnóza
Zatímco Turnerovým syndromem má podobnosti s renální anomálie a vývojové zpoždění, Turnerův syndrom se vyskytuje pouze u žen a často se vyjadřuje jinak. U Turnerova syndromu je nižší výskyt vývojových zpoždění, levostranné srdeční vady jsou konstantní a výskyt renálních abnormalit je mnohem nižší.,
Další RASopathies
- Watson syndrom – Watson Syndrom má řadu podobných charakteristik s Noonan Syndromem, jako je malý vzrůst, plicní chlopně, proměnné, duševní vývoj, a kůži pigmentové změny.
- Cardiofaciocutaneous (CFC) syndrom – CFC syndrom je velmi podobný Noonan Syndromem vzhledem k podobné srdeční a lymfatické vlastnosti. U syndromu CFC je však intelektuální postižení a gastrointestinální problémy často závažnější a výraznější.,
- Costello syndrom podobný syndromu CFC, Costello syndrom má překrývající se rysy s Noonanovým syndromem. Podmínky však lze rozlišit podle jejich genetické příčiny.
- Neurofibromatóza 1 (NF1)
- Williams syndrom
Management
léčba se liší v závislosti na komplikace ale mají tendenci být poměrně standardní, reflexní léčbu z celkové populace., Řízení pokyny, děleno systémy, včetně obecné, vývojové, zubní, růst a krmení, kardiovaskulární, audiologické, hematologické, renální a kosterní, že účet pro opatření, která mají být přijata v době stanovení diagnózy, po stanovení diagnózy, a pokud je symptomatické, byly zveřejněny od Amerického konsorcia.
konkrétně se léčba kardiovaskulárních komplikací podobá léčbě obecné populace a léčba krvácivé diatézy se řídí specifickým nedostatkem faktoru nebo agregací destiček.,
- neuropsychologické testování se doporučuje najít silné stránky a výzvy k přizpůsobení podpory potřebné pro školu a kariéru.
- vzdělávací přizpůsobení, jako je individuální plán vzdělávacího programu, je někdy zapotřebí pro děti ve školním věku.
- logopedie-li řeč a artikulace problémů současnosti
- Fyzikální terapie a pracovní terapie pro hrubé a jemné motoriky zpoždění
- Hypotonie a motorické obtíže často dopad rukopis. Ubytování pro snížení požadavků na rukopis zlepší výkon a ušetří dlouhodobou funkci rukou.,doporučuje se pravidelné sledování a celoživotní sledování abnormalit zjištěných v jakémkoli systému, zejména kardiovaskulárním systému.
Anestezii riziko
i když pár lidí se syndromem Noonanové byly hlášeny k rozvoji maligní hypertermie, gen, mutace chorob je známo, že být spojena s maligní hypertermie je jiný než Noonan syndrom.,
Prognóza
průměrná délka života lidí s Noonan syndromem může být podobné jako v obecné populaci, nicméně, Noonan syndrom může být spojeno s několika zdravotních stavů, které mohou přispět k úmrtnosti. Největším přispěvatelem k úmrtnosti u jedinců s noonanovým syndromem jsou komplikace kardiovaskulárních onemocnění. Prognóza je proto do značné míry závislá na přítomnosti nebo nepřítomnosti srdečního onemocnění, stejně jako na typu a závažnosti onemocnění (pokud je přítomna nemoc)., Zejména noonanův syndrom s hypertrofickou kardiomyopatií je spojen se zvýšenou úmrtností.
Historie
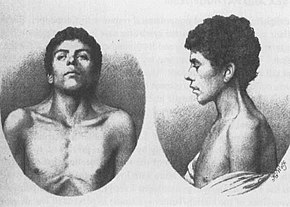
nejstarší známý případ, NS, popsáno v roce 1883 Kobylinski
Jacqueline Noonan praktikoval jako dětský kardiolog na University of Iowa, když si všimla, že děti s vzácný typ srdeční vady, chlopenní vady, plicní stenóza, často měl charakteristický vzhled, s malým vzrůstem, webbed krku, široce rozložené oči, nízko posazené uši., Postiženi byli chlapci i dívky. Tyto charakteristiky byly někdy pozorovány v rodinách, ale nebyly spojeny s hrubými chromozomálními abnormalitami. Studovala 833 lidí se syndromem Noonanové na vrozené onemocnění srdce kliniku, hledá jiné vrozené abnormality, a v roce 1963 prezentoval: „Spojené non-srdeční vady u dětí s vrozenou srdeční vadou“. To popsalo 9 dětí, které kromě vrozené srdeční choroby měly charakteristické rysy obličeje, deformity hrudníku a krátkou postavu.
Dr. John Opitz, bývalý student Dr., Noonanův, nejprve začal nazývat stav „noonanův syndrom“, když viděl děti, které vypadaly jako ty, které popsal Dr. Noonan. Dr. Noonan vyrobila knihu s názvem „Hypertelorismus s Turnerovým Fenotyp“ v roce 1968, kde studovala 19 pacientů, kteří prokázali příznaky svědčící o Noonan Syndromem. V roce 1971 na Symposiu Kardiovaskulárních vad, jméno ‚Noonan syndrom se stal oficiálně uznána.
















Napsat komentář