um doente com…
torsade de pointes
Torsades de Pointes
Eva Buller Viqueiraa, Juana Cabello Pulidob e María José Ibáñez Bulpec
endereço para correspondência
resumo
apresentamos o caso de um homem tratado por convulsões com diazepam em várias ocasiões. Finalmente demonstra-se uma síndrome de QT longo adquirido que o predispunha a torsades de pointes manifestando-se com convulsões e, portanto, tratado erroneamente., A síndrome do QT longo é revisada, descrevendo as diferentes causas congênitas e adquiridas, diagnóstico eletrocardiográfico, medição corrigida do intervalo QT de acordo com a frequência cardíaca e tratamento.
palavras-chave: torsades de Pointes. Síndrome do QT Prolongado. Arritmias Cardíacas.
ABSTRACT
the present is the case of a male treated on several occasions for seizures with diazepam. Eventually, an acquired long QT syndrome was demonstrated which predisposed him to torsades de pointes, manifesting as seizures and henceforth treated erroneously., The long QT syndrome is reviewed, describing the different causes of long QT interval, both congenital and acquired, electrocardiographic diagnosis, according to heart rate and treatment.
Key words: Torsades de Pointes. Long QT Syndrome. Arrhytmias, Cardiac.
introdução
A síndrome do QT longo (SQTL) é uma alteração causada por um alongamento da repolarização do potencial de ação ventricular e predispõe a arritmias malignas tipo torsades de pointes (TdP) ou taquicardia ventricular que podem levar a fibrilação ventricular., Pode ser congênita ou adquirida causada por fármacos, alterações hidroelectrolíticas, jejum ou diversas patologias. Em 1957, Jervell e Lange-Nielsen descreveram o primeiro caso de SQTL familiar com surdez. Selzer e Wray descreveram pela primeira vez o prolongamento do QT e fibrilação ventricular em 1964. Em 1966 Dessertenne descreveu o TdP1, 2.
Caso clínico
apresentamos o caso de um homem de 36 anos, extoxicómano atualmente em tratamento com metadona, fumante e com enolismo crônico., É transexual em tratamento com ciproterona, sofre de transtorno ansioso-depressivo em tratamento com fluoxetina 20 mg, clorazepato dipotássico 50 mg e alprazolam 2 mg. Diagnosticado de infecção por vírus da hepatite C e hamartoma.
o paciente foi encaminhado para urgências hospitalares na suspeita de convulsões tônico-clônicas. A segunda crise foi testemunhada, entrando em parada respiratória que cedeu após ventilação com Ambu. O paciente referia não ter tomado álcool nos dois dias anteriores ao incidente. Fenitoína, pantoprazol e diazepam intravenoso (iv) foram administrados., À chegada ao serviço de urgências hospitalares encontrava-se assintomático e com constantes e exame físico sem nada de interesse. Foram solicitados entre outros testes complementares hemograma com volume corpuscular médio (VCM) alto, bioquímica com hipocalemia, hipocalcemia e hipertransaminassemia (GOT e GPT), tudo em provável relação com seu enolismo crônico. A mioglobina também foi elevada e relacionada a convulsões; positivo para metadona e benzodiazepinas (BZD), resto desinteressante. No eletrocardiograma (ECG) foi registrada uma imagem compatível com bigeminismo., Ele foi administrado ClK iv, pantoprazol iv, propranolol por via oral, hidroloreto de tiamina intramuscular (im), cloridrato de piridoxina iv, e monitoramento eletrocardiográfico foi realizado. A bioquímica foi repetida, mantendo hipocalemia, CK total, CK-Mb e Mb elevados.
foi internado por medicina interna e a bioquímica foi repetida sem incluir potássio. Manteve VCM, GOT, GGT e CK totais elevados. O ECG foi normalizado e o paciente permaneceu assintomático., Ele foi diagnosticado com enolismo crônico, macrocitose, distúrbio do ritmo por hipóxia sem descartar que fosse por BZD e crises convulsivas por privação enólica. Ele foi dispensado mantendo seu tratamento habitual.
No mês seguinte, foi trazido de volta por convulsão, de curta duração e recuperação espontânea. Chegou ao serviço de Urgências assintomático, com um exame sem alterações, VCM elevado, hipocalemia e o ECG apresentava extrasístoles ventriculares isolados. Ele teve alta., No dia seguinte, ele foi novamente derivado de seu centro de saúde por convulsões tônico-clônicas sem privação enólica. Foi mantido em observação e tratado com diazepam iv e registro eletrocardiográfico contínuo. Comentou-se o caso com o cardiologista de plantão, que avaliou o registro com os seguintes achados: bigeminismo ventricular com prolongamento do intervalo QT (0,52 seg.); bigeminismo ventricular e TdP (figura 1); taquicardia ventricular. Foi tratado com ClK iv, sulfato de magnésio iv e monitorização electrocardiográfica., Durante sua internação o controle eletrocardiográfico foi melhorando, apresentou intervalo QT de 0,44 seg., ondas t achatadas e ondas U (sinal de hipocalemia), até chegar a normalizar à alta com um ritmo sinusal a 50 batimentos por minuto (BPM), sem sinais de bloqueio, sem sinais de crescimento de cavidades, com eixo elétrico dentro dos limites da normalidade, sem sinais de isquemia, o intervalo QT de 0,4 seg., ondas t normalizadas e sem presença de onda U (figura 2)., O julgamento clínico à alta foi de síncope por taquicardia ventricular Tipo TdP por QT longo adquirido secundariamente a hipocalemia por etilismo crônico e falta de ingestão. Não necessitou de tratamento específico à alta nem seguimento já que a causa foi adquirida e eliminada.
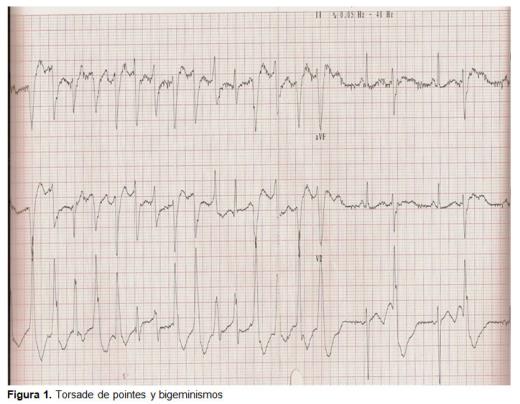

discussão
a síndrome do QT longo pode ocorrer tanto por fatores congênitos quanto por fatores adquiridos., Os congênitos são provocados por alterações genéticas que codificam as proteínas dos canais transmembrana de sódio ou de potássio. Como consequência aumenta a carga positiva no interior da célula por uma falta de saída de sódio ou por um aumento na entrada de potássio. Prolonga-se a repolarização ventricular e, portanto, alonga o intervalo QT, predispondo à TdP. A prevalência é de 1/2000, sendo a síncope a primeira manifestação clínica mais comum. Os pacientes sintomáticos começam com cerca de 12 anos de idade em 50% dos casos e 40 em 90 %., É um dos causadores da Morte súbita infantil; 10% destas mortes apresentam uma mutação genética causadora do SQTL. Numerosos tipos de SQTL congênitos (a síndrome de Romano-Ward subtipos do QTL1-QTL15 e a síndrome de jervell e Lange-Nielson subtipos jln1-JLN2) foram identificados, mas os mais comuns são aqueles incluídos na síndrome de Romano-Ward, e entre eles QTL1-3., Existem fatores predisponentes a eventos cardíacos de acordo com os diferentes tipos de QTL; por exemplo, o QTL-1 é estimulado com atividade física ou estresse emocional (natação), no caso de QTL-2 por estímulos auditivos (como um despertador ou um toque), pós-parto ou estresse emocional. Recentemente, observou-se que a epilepsia é mais comum entre o tipo 2, em 39 %. O diagnóstico diferencial é importante porque muitos são erroneamente tratados como epilepsia pelas convulsões que ocorrem durante a TdP., O diagnóstico do SQTL congênito baseia-se no escore de Schwartz (Tabela 1), que valoriza achados eletrocardiográficos, histórico clínico e familiar. Diagnostica-se de SQTL congênito com um somatório de 3,5 ou mais e sem causas adquiridas. Existem casos de pontuação de 1 a 3 em que o prolongamento do QT está latente, mas pode ser desmascarado realizando um Holter ou séries eletrocardiográficas durante o exercício físico ou teste de epinefrina3,4.,antidepressivos tricíclicos, neurolépticos e procinéticos), interacção medicamentosa, períodos prolongados de jejum ou dietas líquidas proteicas, inibidores enzimáticos (ingestão de sumo de toranja), bradicardia sinusal ou pausas por bloqueio sinoatrial ou atrioventricular, insuficiência hepática ou renal, cardiopatia estrutural (cardiopatia isquémica, insuficiência cardíaca, hipertrofia ventricular), sexo feminino (por possuir um intervalo QT mais longo do que o homem em condições fisiológicas), idade avançada, cardioversão por fibrilação atrial e acidente vascular cerebral agudo5.,
para diagnosticar um SQTL, devemos primeiro saber medir corretamente o intervalo QT no ECG. O intervalo QT compreende desde o início do complexo QRS até o início da onda T, e deve ser medido onde a onda Q Pode ser visualizada (normalmente em DII, V5 ou V6). A medição do QT deve ser ajustada à frequência cardíaca; isso é chamado de QT corrigido (QTc) e é calculado com a fórmula de Bazett.
QTc= QT/√RR
O intervalo RR é a distância entre a onda R e a próxima onda R., Ambos os valores devem ser medidos em milímetros (mm) e passados para segundos antes de serem inseridos na fórmula. Os mm são medidos e multiplicados por 0,04. Em caso de arritmia, várias medições devem ser feitas e a média é calculada. Devemos dar especial atenção a não incluir a onda U na onda T. No caso de não se poder diferenciar bem o final da onda T aconselha-se usar o método da tangente, que consiste em considerar que a onda T termina na interseção da tangente da porção mais inclinada da descendente da onda T e a linha de base (figura 3)., Os valores normais do intervalo QT são distinguidos entre menores de 12 anos e maiores de 12 anos. Nos maiores de 12 anos diferencia-se entre homens e mulheres, considerando-se normal entre 0,39 e 0,45 nos primeiros e entre 0,39 e 0,46 nas segundas3.

o prolongamento do intervalo QT pode predispor a uma torção de pontas ou TdP. A TdP é uma variação polimórfica irregular do QRS que vai mudando de positivo para negativo para começar de novo; parece que as pontas se torcem ao redor do eixo elétrico., A frequência cardíaca é entre 150 e 300 BPM com intervalos RR irregulares. Começa subitamente e não é sustentada.
O tratamento da TdP é cardioversão em caso de comprometimento hemodinâmico. Todos os medicamentos que possam provocar o prolongamento do intervalo QT devem ser retirados. O sulfato de magnésio é de primeira escolha seja congênito ou adquirido e independente do nível de magnésio no soro: um bolo de 2 g a passar em 2-3 minutos seguido de infusão iv 2-4 mg/min e podendo passar um segundo bolo se a TdP se repetir enquanto se está colocando a infusão IV., É compatível com a gravidez. Pode ocorrer hipertensão arterial e asistolia. O antídoto para o sulfato de magnésio é o gluconato de cálcio. O isoproterenol é usado quando o sulfato de magnésio e o pacemaker falham, ou a equipe treinada não é treinada para colocar o pacemaker. Não deve ser usado no SQTL congênito. Como efeitos colaterais são comuns palpitações, rubor facial e calor. Quanto aos níveis séricos de potássio, demonstrou-se que é preferível que estejam nos limites mais elevados do que o normal, podendo requerer a sua administração IV., Não está contra-indicado na gravidez e nunca deve ser administrado em bolus, porque pode produzir arritmias e depressão cardíaca, provocando a morte. Deve ser monitorizado com ECG. O pacemaker temporário endovenoso pode ser colocado a cerca de 100 bpm se o magnésio não conseguir evitar o TdP.
O tratamento a longo prazo do SQTL no caso de ser adquirido não é necessário, porque uma vez que a causa é eliminada geralmente cede. Em vez disso, se congênito é aconselhável o uso de β-bloqueadores por via oral ao diminuir de 0,97 a 0,31 os eventos cardíacos por paciente e ano., É aconselhável manter uma frequência cardíaca durante o exercício inferior a 130 BPM. Os suplementos de potássio são recomendados no QTL-2. O desfibrilador implantável e a denervação simpática cardíaca esquerda são recomendados em pacientes com história de parada cardíaca, clínica antes do ano de vida ou JLN-1. A educação é importante para evitar estímulos precipitantes (exercício físico, natação, falta de sono, estímulos auditivos, estímulos simpáticos intensos, dor, sofrimento, raiva surdos)3,6-8.,
parece-nos que a medição do QTc é algo negligenciado na atenção primária apesar de ser fácil de calcular e muito acessível, já que o ECG é um teste complementar muito comum em nosso trabalho diário. O simples facto de o reconhecer e agir em conformidade pode prevenir graves consequências. A medição sistemática do intervalo QT para sua detecção precoce em tratamento com medicação que o alonga poderia ser recomendada, assim como nas alterações eletrolíticas.
Conflito de interesses
Os autores declaram não ter nenhum conflito de interesses.,
Bibliografia
1. Kallergis em, Goudis CA, SIMANTIRAKIS EN, KOCHIADAKIS GE, vardas PE. Mechanisms, risk factors, and management of acquired long QT Syndrome: a comprehensive review. Scientific World Journal. 2012; 2012: 212178.
3. Muñoz Castelhano J. síndrome de QT largo e Torsade de Pointes. Emergências. 2004; 16: 85-92.
















Deixe uma resposta