introdução
deficiência intelectual (ID) afeta quase 1-2% da população e é a doença de desenvolvimento neurológico mais comum (NDD). Um número substancial de pacientes ID são encontrados para ter uma causa genética (revisado em Bessa et al., 2012)., As técnicas de análise genômica atualmente utilizadas para a investigação da etiologia muitas vezes levam à identificação de variantes muito raras, quase privadas, sendo a coleta de pacientes com alterações no mesmo gene um aspecto crucial da definição de uma nova entidade clínica.
no início deste ano, foram descritos doentes com mutações no gene EBF3, apresentando uma síndrome de desenvolvimento neurológico incluindo ID, ataxia, hipotonia, dismorfismos Faciais ligeiros e anomalias genitourinárias (OMIM 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., O gene EBF3 (Early B cell Factor 3) codifica um membro da família altamente conservada do factor de transcrição do factor B, expressa em altos níveis no desenvolvimento do sistema nervoso (dados obtidos do Portal GTEx). EBF3 é um alvo transcritional da ARX, e mostrado para ser regulado por NeuroD e ARX (Friocourt e Parnavelas, 2011). A ARX codifica um fator de transcrição crítico para o desenvolvimento embrionário que, por muitos anos, tem sido associado a uma ampla gama de distúrbios de desenvolvimento neurológico., A insuficiência intelectual, o sistema nervoso central e as anomalias genitourinárias observadas em pacientes com mutações no EBF3 e no ARX podem refletir a contribuição de ambas as proteínas para os mesmos processos moleculares e celulares (Chao et al., 2017). A função EBF3 também foi estudada em modelos animais. A ablação de seus ortólogos em minhocas e moscas leva à diminuição do desenvolvimento neuronal (Prasad et al., 1998; Hattori et al., 2013)., Em ratos, nocautear o Ebf3 leva à letalidade neonatal e defeitos de migração neuronal, com falha do projeto de neurônios olfativos para o bulbo olfativo dorsal (Wang et al., 2004). Os exatos mecanismos patogênicos de EBF3 mutações ainda não está totalmente elucidado, mas o tipo de variantes descritas até agora sugerem que haploinsufficiency, ganho de função, e negativo dominante são possíveis mecanismos patogênicos para as variantes descritas (Chao et al., 2017; Sleven et al., 2017).,
neste trabalho contribuímos com um paciente com a menor deleção (600 Kb) relatada até à data afetando a totalidade do gene EBF3 e com uma apresentação clínica sobrepondo-se a de pacientes com variantes nucleotídicas únicas EBF3 (SNVs). Além disso, fazemos uma comparação clínica dos pacientes com supressões terminais 10q previamente publicados e relatamos que, apesar das diferenças de tamanho, há uma sobreposição fenotípica significativa entre os pacientes com essas alterações., Estes resultados acrescentam ao conhecimento actual das perturbações relacionadas com o EBF3 e suportam a haploinsuficiência do EBF3 como chave na síndrome de desenvolvimento neurológico associada a supressões de 10qter.materiais e Métodos o doente foi verificado num grande estudo de distúrbios do desenvolvimento neurológico em Portugal, no qual o registo dos doentes e das famílias foi feito pelo médico de referência, a informação clínica foi recolhida numa base de dados anónima de acordo com a autoridade Portuguesa de protecção de Dados (CNPD) e foi obtido consentimento informado por escrito para todos os participantes., O consentimento informado para o presente paciente foi fornecido pela mãe para o estudo genético e publicação de Resultados (incluindo fotos). O estudo foi aprovado pelo Comitê de Ética do centro de Genética Médica Dr. Jacinto Magalhães, Instituto Nacional de Saúde Dr. Ricardo Jorge.o ADN genómico foi extraído do sangue periférico utilizando um kit de isolamento de ADN Citogene® (Citomed, Portugal). o aCGH foi realizado usando Agilent 180 K array (AMADID:023363) contra uma referência diplóide de DNA (DNA de referência MegaPoll de Kreatech, Kreatech Diagnostics, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., A análise foi realizada em uma máquina PCR em tempo Real de 7500-rápido (Applied Biosystems, Foster City, CA, EUA) usando Power SYBR Green® (Applied Biosystems, Foster City, CA, EUA) de acordo com as recomendações do fabricante e seguindo as diretrizes gerais para qPCR. A especificidade de cada reação foi verificada pela geração de uma curva de fusão para cada um dos fragmentos amplificados. A eficiência da iniciadora foi calculada através da geração de uma curva padrão ajustando a percentagem de eficiência normal aceita (iniciadores utilizados listados em dados suplementares)., Os valores de Ct obtidos para cada teste foram analisados em software DataAssist™ (Applied Biosystems, Foster City, CA, EUA).
resultados
Aqui descrevemos um doente com uma deleção de novo que afecta o EBF3. O paciente é uma menina de 11 anos com ID grave (quociente de desenvolvimento global = 27 aos 7 anos de idade), nascida de pais não consanguíneos e sem história familiar de distúrbios do desenvolvimento neurológico. Ela nasceu após uma gravidez biamniótica bicoriônica (sua irmã sendo saudável), por parto vaginal, às 35 semanas de gestação. Os parâmetros de nascimento foram: peso, 1.830 g (P3); Comprimento, 42.,5 cm (P10); e OFC, 30,6 cm (P10), com uma pontuação Apgar de 8/9 (1 e 5 min, respectivamente). O período neonatal foi complicado com a sépsis e o diagnóstico de esferocitose hereditária (herdada de sua mãe). O atraso no desenvolvimento Global foi observado nos primeiros meses, com controle de cabeça alcançado aos 12 meses, sentado aos 18 meses, caminhada independente aos 30 meses, e nenhuma palavra falada aos 3 anos de idade., Teve pielonefrite aos 19 meses (o ultra-som renal não mostrou anomalias), refluxo gastroesofágico e otite média recorrente, com perda auditiva condutiva que requereu intervenção cirúrgica e um aparelho auditivo. Suspeitava-se de epilepsia aos 5 meses (episódios de actividade suspensa), mas o EEG estava normal.foi observada pela primeira vez com a idade de 3 anos e 5 meses (figuras 1A,B), altura em que apresentou hipotonia muscular, face hipotónica, estrabismo e sensibilidade reduzida à dor., Ela também apresentou leve dysmorphic características (Figura 1A): rosto triangular, pequena baixa das orelhas, com destaque anti-hélice, sobrancelhas arqueadas, anteversão narinas, bulbosa ponta nasal, boca pequena com downturned cantos, apontou o queixo, pescoço curto, e proeminente dedo fetal almofadas, bem como uma leve baixa estatura (89 cm, correspondentes a cerca de 2SD).
Figura 1. A) aspecto Facial do doente aos 3 anos e 5 meses, mostrando as orelhas pequenas e de baixo nível com antielix e B) protecções fetais nos dedos., C) aspecto Facial do doente aos 11 anos de idade. (D) Destacadas em cinza a 600 Kb eliminação de 10q26.3 região; um zoom do EBF3 gene na DGV banco de dados revela a existência de 3 eliminações em 3 controles que afetam o primeiro exon 6 do EBF3 (NM_001005463); CNVs dentro desta região encontrado no controle de populações incluem exclusões nvs825626 (presente em 1/31 indivíduos), nvs552315 (presente em 1/17421 indivíduos) e nsv552316 (presente em 1/31 indivíduos). E) a representação esquemática das transcrições EBF3.,
ressonância magnética cerebral foi realizada aos 6 anos, mas não foram observadas anomalias. Com a idade de 10 anos, ela foi reavaliada; ela ainda tinha otite média recorrente, mas de outra forma estava em boa saúde global. A língua era muito pobre (duas frases de palavras faladas após 8 anos). Ela teve problemas de comportamento, com movimentos estereotipados (movimentos rotativos, mastigação de roupas, retropulsão na cabeça), pontuação para transtorno grave do espectro do autismo (ADI-R e ADO) aos 7 anos; ela mostrou agitação e comportamento agressivo (auto e hetero) e foi medicada com drogas antipsicóticas., Uma cirurgia ortopédica foi realizada para pes planus. As características faciais eram semelhantes às descritas anteriormente, com incisivos centrais superiores espaçados( figura 1C); ela tinha óculos para estrabismo e hipermetropia.
a Análise do DNA genômico por aCGH revelou de novo de 600 kb eliminação de 10p26.3 (Figura 1D) afetando três genes—MGMT (codificação da enzima O-6-methylguanine-DNA metiltransferase, envolvidos no reparo de DNA), EBF3 e GLRX (codificação glutaredoxin, uma pequena thioltransferase que remove proteínas GSH adducts), de que EBF3 era mais provável que a doença associada gene.,
discussão
o paciente apresentado foi analisado pela primeira vez pelo aCGH há alguns anos. No momento da análise do aCGH, a existência das três variantes presentes na Base de dados de variantes genômicas (DGV)1 afetando os primeiros cinco exons do gene EBF3 (figura 1E; Park et al., 2010; Cooper et al., 2011), bem como a ausência de outras doenças conhecidas que causam mutações neste gene, nos levam a classificá-lo como uma variante de significado desconhecido (VOUS). No entanto, as publicações recentes de mutações EBF3 (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) e as semelhanças clínicas com os casos relatados, nos levam a reavaliar a variante e nos fazem acreditar que a exclusão do EBF3 pode de fato estar contabilizando a doença no paciente. Um dos aspectos que levantou dúvidas sobre a patogenicidade desta variante, em primeiro lugar, foi a existência de população controles de rolamento de eliminações dos primeiros seis exões do gene, em heterozigose (dados obtidos a partir de DGV base de dados de fevereiro de 2017) (Figura 1D). Mesmo que uma transcrição do EBF3 começando em Exon13b está listada na Base de dados Ensembl (ENST00000440978.,1) (Figura 1E), o que poderia explicar como a exclusão do primeiro exon pode, eventualmente, resultar em um fenótipo normal, esta transcrição, exclui-se o DNA domínio de ligação de EBF3, e seu padrão de expressão e relevância funcional não foram caracterizados. Após reavaliação, no entanto, os CNVs descritos por Cooper e colegas (nsv552315 e nsv552316) (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) foram considerados como estando no limiar de detecção pelo microarray SNP e não podem ser a base para a exclusão de um gene candidato, particularmente à luz da forte evidência genética e funcional para a relevância das mutações EBF3 para a doença (Evan Eichler, Greg Cooper e Bradley Coe, comunicação pessoal).,
nosso paciente mostra muitas semelhanças clínicas com pacientes previamente descritos com mutações no EBF3( 21 casos resumidos na Tabela 1), tais como atraso no desenvolvimento global, fala expressiva retardada, hipotonia, aumento do limiar da dor, problemas comportamentais e características faciais (face longa/triangular, testa grande, face hipotônica)., No entanto, mesmo que nosso paciente tenha tido atraso significativo no desenvolvimento motor, não foi detectada ataxia significativa (com algumas limitações no exame clínico, como a criança não foi cooperativa), e não houve anomalias cerebelares na ressonância magnética cerebral.

Quadro 1. Comparação clínica do presente caso com os casos notificados com mutações pontuais/indels no gene EBF3.
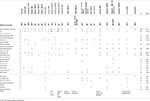
Tabela 2., Comparação clínica do presente caso com os casos notificados com supressões que afectam a citobanda 10q26.
O EBF3 variantes descritas na literatura no início de 2017 incluem mutações de ponto previsto para ser deletérios e pequenas inserções e exclusões principais no quadro de exclusão da chave de aminoácidos ou frameshift, previsivelmente causar início de truncamento da proteína resultante ou nonsense-mediated decay., As mutações foram concentradas em partes do gene que codificava o domínio de ligação ao ADN do EBF3, e foram previstas através de diferentes métodos para levar a uma perda de função deste factor de transcrição, sugerindo assim uma função reduzida e haploinsuficiência como o mecanismo subjacente à perturbação do desenvolvimento neurológico nestes doentes. Ratos noctívagos para o Ebf3 são descritos para apresentar letalidade neonatal e defeitos de migração neuronal, com falha dos neurônios olfativos para projetar para o bulbo olfativo dorsal (Wang et al.,, 2004), mas nenhuma Descrição é feita de um fenótipo nos animais heterozigóticos, que são realmente apresentados como controles em muitos dos experimentos, não suportando assim o modelo de haploinsuficiência. Nós fizemos esforços para obter e estudar o fenótipo de desenvolvimento neurológico destes animais, mas não foram bem sucedidos, como a linha de knock-out Ebf3(o/E2) pode ter sido descontinuada (Joseph W. Lewcock, comunicação pessoal). No entanto, o caso atual, juntamente com os pacientes resumidos na Tabela 2, apoiam a hipótese de HAPLOINSUFICIÊNCIA EBF3 como causa da doença.,
em resumo, a descrição Actual reforça a perda de função/haploinsuficiência do EBF3 como causa da doença do desenvolvimento neurológico, e reforça a associação deste gene a uma síndrome clínica característica dentro deste espectro.
contribuições do autor
FL realizou os estudos moleculares e analisou os dados moleculares. MG e GS recolheram dados clínicos. FL, JP, and PM reviewed all EBF3 mutation cases in the literature. FL, GS, JP e PM redigiram o jornal. A PM obteve financiamento para este estudo. O estudo foi realizado sob a direcção da PM.,(PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Este artigo foi desenvolvido no âmbito do projecto NORTE-01-0145-FEDER-000013, apoiado pelo Programa Operacional Regional do Norte de Portugal (NORTE 2020), ao abrigo do Acordo de Parceria Portugal 2020, através do Fundo Europeu de Desenvolvimento Regional (FEDER).
Declaração de conflito de interesses
JP foi empregado pela empresa CGC Genetics.,os outros autores declaram que a pesquisa foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.Agradecemos à paciente e sua família pela participação nos estudos genéticos e por permitir esta publicação. Estamos gratos à Dra. Ana Maria Fortuna e à Dra. Margarida Reis Lima por viabilizarem este projecto e por facilitarem a nossa colaboração com a CGM.
notas
Friocourt, G., and Parnavelas, J. (2011). A identificação das metas da Arx revela novos candidatos para controlar a migração e diferenciação do interneuron cortical. Frente. Celula. Neuroci. 5:28. doi: 10.3389 / fncel.2011.00028
PubMed Abstract / CrossRef Full Text | Google Scholar
















Deixe uma resposta