はじめに
知的障害(ID)は、人口のほぼ1-2%に影響を与え、最も一般的な神経発達障害(NDD)である。 相当数のID患者が遺伝的原因を有することが見出される(bessa et al., 2012)., 病因の調査に現在使用されているゲノムワイド解析技術は、しばしば非常にまれなほとんどプライベートバリアントの同定につながり、同じ遺伝子の変化を有する患者のコレクションは、新しい臨床実体の定義の重要な側面である。
今年初め、EBF3遺伝子に変異を有する患者が記載されており、ID、運動失調、低血圧、軽度の顔面異形、および尿生殖器異常を含む神経発達症候群(OMIM617330)(Chao et al.,2017;Harms et al.,2017;Sleven et al., 2017)., EBF3(初期B細胞因子3)遺伝子は、高度に保存された初期B細胞因子転写因子ファミリーのメンバーをコードし、発達中の神経系(GTExポータルから取得されたデータ)で高レベルで発現する。 EBF3はARXの転写標的であり、NeuroDおよびARXによって調節されることが示されている(FriocourtおよびParnavelas、2011)。 ARXは、長年にわたり、神経発達障害の広い範囲に関連付けられている、胚発生のための重要な転写因子をコードしています。, EBF3およびARXの両方の変異を有する患者において観察された知的障害、中枢神経系および尿生殖器異常は、同じ分子および細胞プロセスに対する両タンパク質の寄与を反映している可能性がある(Chao et al., 2017). EBF3機能はまた動物モデルで調査されました。 虫およびハエにおけるそのオルソログのアブレーションは、神経発達の障害をもたらす(Prasad et al.,1998;Hattori et al., 2013)., マウスでは、Ebf3をノックアウトすると、新生児致死率および神経遊走欠陥につながり、嗅覚ニューロンが背側嗅球に投影されなくなる(Wang et al., 2004). EBF3変異の正確な病原性メカニズムはまだ完全には解明されていないが、これまでに記載された変異体のタイプは、ハプロイン不足、機能の獲得、およびドミナントネガティブが記載された変異体の病原性メカニズムである可能性があることを示唆している(Chao et al.,2017;Sleven et al., 2017).,
この作業では、ebf3遺伝子の全体に影響を与えるこれまでに報告された最小の削除(600キロバイト)を持つ患者とEBF3一塩基変異体(SNVs)を持つ患者の さらに、我々は、以前に公開された大きなターミナル10q欠失を有する患者の臨床的比較を行い、サイズの違いにもかかわらず、これらの変化を有する患者間の有意な表現型の重なりがあることを報告する。, これらの知見は、EBF3関連疾患の現在の知識に追加し、EBF3ハプロイン不足を10qter欠失に関連する神経発達症候群の鍵としてサポートしています。
材料と方法
患者は、患者と家族の登録は、参照医師によって行われたポルトガルの神経発達障害の大規模な研究内で確認された、臨床情報は、ポルトガルのデータ保護庁(CNPD)に従って匿名のデータベースに収集され、すべての参加者のために書面によるインフォームドコンセントが得られた。, 現在の患者のためのインフォームドコンセントは、遺伝子研究と結果(写真を含む)の公表のために母親によって提供されました。 この研究は、医学遺伝学センターの倫理委員会Jacinto Magalhães博士、国立衛生研究所のRicardo Jorge博士によって承認されました。Citogene(登録商標)DNA isolation kit(Citomed,Poltual)のいずれかを用いて末梢血からゲノムDNAを抽出した。 aCGHは、二倍体DNA参照(Kreatech’S MegaPoll参照DNA、Kreatech Diagnostics、Amsterdam)に対してAgilent180Kアレイ(AMADID:023363)を用いて行った。, aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., 分析は、Power SYBR Green(商標)(Applied Biosystems、Foster City、CA、USA)を使用して、7500高速リアルタイムPCRマシン(Applied Biosystems、Foster City、CA、USA)で、製造業者の推奨に従い、qPCRに関する一般的なガイドラインに従っ 各反応の特異性は、増幅された断片のそれぞれについての融解曲線の生成によって検証された。 プライマー効率は、受け入れられた正常効率率(補足データに記載されている使用されるプライマー)に適合する標準曲線の生成によって計算された。, 各試験について得られたct値を、Dataassisttmソフトウェア(Applied Biosystems,Foster City,CA,USA)で分析した。
結果
ここでは、EBF3に影響を与えるde novo削除を有する患者について説明します。 患者は、重度のID(グローバル開発商=27歳で7歳)を持つ11歳の少女であり、非血族的な両親から生まれ、神経発達障害の家族歴がない。 彼女は妊娠35週で、膣分娩によって、biamniotic bichorionic双子の妊娠(彼女の妹は健康である)の後に生まれました。 出生パラメータは、体重、1,830g(P3)、長さ、42であった。,5cm(P10);およびOFC、30.6cm(P10)、Apgarスコアは8/9(それぞれ1分および5分)であった。 新生児期は敗血症および遺伝性球状赤血球症(母親から継承)と診断された。 最初の数ヶ月でグローバルな発達遅延が認められ、頭部制御は12ヶ月で達成され、18ヶ月で座って、30ヶ月で独立した歩行、3歳で話された言葉はなかった。, 彼女は19ヶ月で腎盂腎炎(腎超音波は異常を示さなかった)、胃食道逆流および再発性中耳炎を有し、外科的介入および補聴器を必要とする伝導性難聴 てんかんは5ヶ月(活動停止のエピソード)で疑われたが、脳波は正常であった。
彼女は3歳5ヶ月で最初に観察された(図1A、B)、その時点で彼女は筋緊張低下、低張顔面、斜視、および痛みに対する感受性の低下を示した。, 彼女はまた、軽度の異形性の特徴(図1A)を提示した:三角形の顔、顕著な反らせんを有する小さな低セット耳、アーチ状の眉毛、前傾鼻、球根状の鼻先、下向きの角を有する小さな口、尖った顎、短い首、および顕著な指胎児パッド、ならびに軽度の低身長(89cm、約2SDに相当)。

図1. (A)顕著な反螺旋および(B)指の胎児のパッドが付いている小さく、低セットの耳を示す3年および5か月の患者の顔の出現。, (C)11歳の患者の顔の外観。 (D)グレーで強調表示された600Kbの削除10q26.3領域;DGVデータベース内のEBF3遺伝子のズームインは、3EBF3の最初の6エクソンに影響を与えるコントロールでの欠失の存在を明らかにする(NM_001005463);コントロール集団で見つかったこの領域内のCnvは、欠失nvs825626(1/31個人に存在)、nvs552315(1/17421個人に存在)とnsv552316(1/31個人に存在)を含む。 (E)EBF3転写産物の概略図。,
脳MRIは6年で行われたが、異常は認められなかった。 10歳の時に再評価され、まだ再発性中耳炎を患っていたが、それ以外の場合は良好なグローバルヘルスであった。 言語は非常に悪かった(8年後に話された二つの単語の文章)。 彼女は行動の問題を抱えていました,ステレオタイプの動きで(回転運動,服を噛む,頭のretropulsion),重度の自閉症スペクトラム障害のための得点(ADI-RとADOS)7年;彼女は攪拌と積極的な行動を示しました(自動とヘテロ)そして、抗精神病薬で薬を投与されました., Pesへん平に対して整形外科手術を行った。 顔の特徴は、前に説明したものと同様であり、上部中切歯が間隔をあけていた(図1C)。
aCGHによるゲノムDNAの解析により、de novo600kbの欠失が10p26.3で明らかになった(図1D)MGMT(DNA修復に関与する酵素O—6-メチルグアニン-DNAメチルトランスフェラーゼをコードする)、EBF3およびGLRX(グルタレドキシン、タンパク質GSH付加体を除去する小さなチオルトランスフェラーゼをコードする)、EBF3が最も可能性の高い疾患関連遺伝子であった。,
ディスカッション
提示された患者は、数年前にaCGHによって最初に分析されました。 ACGH解析の時点で、ゲノム変異体(DGV)1のデータベースに存在する三つの変異体の存在は、EBF3遺伝子の最初の五つのエクソンに影響を与える(図1E;Park et al.,2010;Cooper et al.,2011)、ならびにこの遺伝子に突然変異を引き起こす他の既知の疾患がないことは、それを未知の意義の変種(VOUS)として分類することを導く。 しかしながら、EBF3変異の最近の刊行物(Chao et al.,2017;Harms et al.,2017;Sleven et al.,、2017)および報告された症例との臨床的類似性により、バリアントを再評価し、EBF3欠失が実際に患者の疾患を占めている可能性があると信じさせる。 そもそもこの変異の病原性に疑問を投げかけた側面の一つは、この遺伝子の最初の六つのエクソンの欠失を有する集団制御がヘテロ接合性において存在することであった(2017年1月現在のDGVデータベースから取得されたデータ)(図)。 EBF3から始まるExon13bのトランスクリプトがEnsemblデータベースにリストされているにもかかわらず(ENST00000440978。,1)(図1E)は、最初のエクソンの欠失が最終的に正常な表現型をもたらすことができる方法を説明することができ、この転写産物はEBF3のDNA結合ドメインを除外し、その発現パターンと機能的関連性は特徴付けられていない。 しかしながら、再評価の際に、Cooperらによって記載されたCnv(nsv552315およびnsv552316)(Chao et al.,2017;Harms et al.,2017;Sleven et al.,,2017)は、SNPマイクロアレイによる検出の閾値であると考えられており、特にEBF3変異と疾患との関連性に関する強力な遺伝的および機能的証拠に照らして、候補遺伝子の排除の基礎となることはできない(Evan Eichler,Greg Cooper and Bradley Coe,personal communication)。,
私たちの患者は、EBF3の突然変異を有する以前に記載された患者と多くの臨床的類似性を示しています(21例は表1にまとめられています)、グローバルな発達遅延、表現力遅延、低血圧、疼痛閾値の増加、行動上の問題および特徴的な顔の特徴(長い/三角形の顔、大きな額、低張性の顔)など。, しかし,運動発達に著しい遅延があったにもかかわらず,有意な運動失調は検出されず(小児は協調性ではなかったため,臨床検査にはいくつかの制限があった),脳MRIには小脳異常はなかった。

テーブル1. EBF3遺伝子における点突然変異/インデルを有する報告された症例と本症例の臨床的比較。
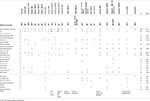
テーブル2., 10q26サイトバンドに影響を与える欠失を有する報告された症例と本症例の臨床的比較。
文献に記載されているEBF3変異体には、2017年の初めに有害であると予測される点突然変異が含まれ、キーアミノ酸のフレーム内欠失またはフレームシフトにつながる小さな挿入および欠失が含まれ、結果として生じるタンパク質の早期切断またはナンセンス媒介崩壊を引き起こすことが予想される。, 変異はEBF3のDNA結合ドメインをコードする遺伝子の一部に集中しており、この転写因子の機能の喪失につながるさまざまな方法によって予測され、これらの患者の神経発達障害の根底にあるメカニズムとして機能の低下とハプロイン不足を示唆している。 Ebf3のノックアウトマウスは、背側ol球に投影する嗅覚ニューロンの障害を伴う、新生児致死および神経遊走欠陥を提示するために記載されている(Wang et al.,,2004)、しかし、実際には実験の多くでコントロールとして提示されているヘテロ接合動物の表現型の記述はなされておらず、したがって、ハプロイン不足モデルをサポートしていない。 我々は、これらの動物の神経発達表現型を取得し、研究するための努力をしたが、Ebf3(O/E2)ノックアウトラインが中止されている可能性があるため、成功しませんでした(Joseph W.Lewcock、パーソナルコミュニケーション)。 しかしながら、現在の症例を表2にまとめた患者とともに、原因疾患としてのEBF3ハプロイン不足の仮説を支持している。,
要約すると、現在の説明は、神経発達疾患の原因としての機能/ハプロイン不足のEBF3損失を強化し、このスペクトル内の特徴的な臨床症候群とこの
著者の貢献
FLは、分子研究を行い、分子データを分析しました。 MGおよびGSは臨床データを収集した。 FL、JP、およびPMは、文献におけるすべてのEBF3変異症例をレビューした。 FL、GS、JP、およびPMが論文を起草しました。 PMはこの研究のための資金を得ました。 この研究はPMの指示の下で行われた。,
資金調達
FCT-Fundação para A Ciúncia e A Tecnologiaプロジェクトや奨学金内(PIC/IC/83026/2007,PIC/IC/83013/2007,SFRH/BD/90167/2012). この記事は、プロジェクトノルテの範囲の下で開発されています-01-0145-FEDER-000013は、欧州地域開発基金(FEDER)を通じて、ポルトガル2020パートナーシップ協定の下で、北ポルトガル地域運営プログラム(NORTE2020)によってサポートされています。
利益相反声明
JPはCGC Genetics社に採用されました。,
他の著者は、この研究は、潜在的な利益相反と解釈され得る商業的または財政的関係がない場合に行われたと宣言している。
謝辞
私たちは、遺伝子研究への参加とこの出版物を許可するために、患者と彼女の家族に感謝したいと思います。 このプロジェクトを可能にし、CGMとのコラボレーションを促進してくれたAna Maria Fortuna博士とMargarida Reis Lima博士に感謝しています。
脚注
1., ^http://dgv.tcag.ca/dgv/app/homeで利用可能なゲノム変異体のデータベース。
Friocourt,G.,And Parnavelas,J.(2011). Arxターゲットの同定は、皮質介在ニューロンの移行と分化を制御するための新しい候補を発表します。 フロント セル ニューロシティ 5:28. ドイ:10.3389/2011.00028
PubMed Abstract/CrossRef Full Text|Google Scholar
















コメントを残す