UN PACIENTE CON…
Torsade de pointes
Torsades de Pointes
Eva Buller Viqueiraa, Juana Cabello Pulidob y María José Ibáñez Bulpec
Dirección para correspondencia
RESUMEN
Presentamos el caso de un varón tratado por convulsiones con diazepam en varias ocasiones. Finalmente se demuestra un síndrome de QT largo adquirido que le predisponía a torsades de pointes manifestándose con convulsiones y, por tanto, tratado erróneamente., Se repasa el síndrome del QT largo, describiendo las diferentes causas tanto congénitas como adquiridas, diagnóstico electrocardiográfico, la medición corregida del intervalo QT según la frecuencia cardiaca y tratamiento.
Palabras clave: Torsades de Pointes. Síndrome de QT Prolongado. Arrtimias Cardiacas.
ABSTRACT
The present is the case of a male treated on several occasions for seizures with diazepam. Eventually, an acquired long QT syndrome was demonstrated which predisposed him to torsades de pointes, manifesting as seizures and henceforth treated erroneously., The long QT syndrome is reviewed, describing the different causes of long QT interval, both congenital and acquired, electrocardiographic diagnosis, according to heart rate and treatment.
Key words: Torsades de Pointes. Long QT Syndrome. Arrhytmias, Cardiac.
Introducción
El síndrome del QT largo (SQTL) es una alteración causada por un alargamiento de la repolarización del potencial de acción ventricular y predispone a arritmias malignas tipo torsades de pointes (TdP) o taquicardia ventricular que pueden desembocar en fibrilación ventricular., Puede ser congénita o adquirida causado por fármacos, alteraciones hidroelectrolíticas, ayuno o distintas patologías. En 1957 Jervell y Lange-Nielsen describieron el primer caso de SQTL familiar con sordera. Selzer y Wray describieron por primera vez la prolongación del QT y fibrilación ventricular en 1964. En 1966 Dessertenne describió la TdP1,2.
Caso clínico
Presentamos el caso de un varón de 36 años, extoxicómano actualmente en tratamiento con metadona, fumador y con enolismo crónico., Es transexual en tratamiento con ciproterona, padece de trastorno ansioso-depresivo en tratamiento con fluoxetina 20 mg, clorazepato dipotásico 50 mg y alprazolam 2 mg. Diagnosticado de infección por virus de hepatitis C y hamartoma.
El paciente fue derivado a urgencias hospitalarias ante la sospecha de convulsiones tónico-clónicas. La segunda crisis fue presenciada, entrando en parada respiratoria que cedió tras ventilación con ambú. El paciente refería no haber tomado alcohol en los dos días previos al incidente. Se administró fenitoína, pantoprazol y diazepam intravenoso (iv)., A la llegada al servicio de urgencias hospitalarias se encontraba asintomático y con constantes y exploración física sin nada de interés. Se solicitaron entre otras pruebas complementarias hemograma con un volumen corpuscular medio (VCM) alto, bioquímica con hipopotasemia, hipocalcemia e hipertransaminasemia (GOT y GPT), todo en probable relación con su enolismo crónico. La mioglobina también estaba elevada y se relacionó con las convulsiones; positivo para metadona y benzodiacepinas (BZD), resto sin interés. En el electrocardiograma (ECG) se registró una imagen compatible con bigeminismo., Se le administró ClK iv, pantoprazol iv, propranolol por vía oral, hidroloruro de tiamina intramuscular (im), clorhidrato de piridoxina iv, y se realizó monitorización electrocardiográfica. Se repitió la bioquímica, manteniendo hipopotasemia, CK total, CK-Mb y Mb elevados.
Se ingresó a cargo de medicina interna y se volvió a repetir la bioquímica sin incluir potasio. Mantenía VCM, GOT, GGT y CK totales elevados. El ECG se normalizó y el paciente se mantenía asintomático., Se le diagnosticó de enolismo crónico, macrocitosis, trastorno del ritmo por hipoxia sin descartar que fuera por BZD y crisis convulsivas por deprivación enólica. Se le dio el alta manteniendo su tratamiento habitual.
Al mes siguiente fue traído de nuevo por convulsión de breve duración y recuperación espontánea. Llegó al servicio de urgencias asintomático, con una exploración sin alteraciones, VCM elevado, hipopotasemia y el ECG presentaba extrasístoles ventriculares aislados. Fue dado de alta., Al día siguiente fue derivado de nuevo desde su centro de salud por convulsiones tónico-clónicas sin deprivación enólica. Se mantuvo en observación y se trató con diazepam iv y registro continuo electrocardiográfico. Se comentó el caso con el cardiólogo de guardia, quien valoró el registro con los siguientes hallazgos: bigeminismo ventricular con prolongación del intervalo QT (0,52 seg.); bigeminismo ventricular y TdP (figura 1); taquicardia ventricular. Se trató con ClK iv, sulfato magnésico iv y monitorización electrocardiográfica., Durante su ingreso el control electrocardiográfico fue mejorando, presentó un intervalo QT de 0,44 seg., ondas T aplanadas y ondas U (signo de hipopotasemia), hasta llegar a normalizarse al alta con un ritmo sinusal a 50 latidos por minuto (lpm), sin signos de bloqueo, sin signos de crecimiento de cavidades, con eje eléctrico dentro de los límites de la normalidad, sin signos de isquemia, el intervalo QT de 0,4 seg., ondas T normalizadas y sin presencia de onda U (figura 2)., El juicio clínico al alta fue de síncope por taquicardia ventricular tipo TdP por QT largo adquirido secundariamente a hipopotasemia por etilismo crónico y falta de ingesta. No requirió de tratamiento específico al alta ni seguimiento puesto que la causa fue adquirida y se eliminó.
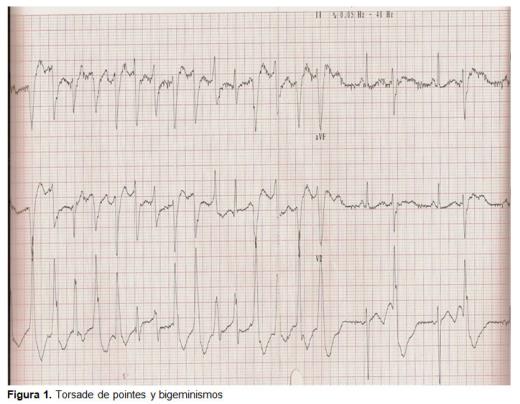

Discusión
El síndrome del QT largo se puede producir tanto por factores congénitos como por factores adquiridos., Los congénitos son provocados por alteraciones genéticas que codifican las proteínas de los canales transmembrana de sodio o de potasio. Como consecuencia aumenta la carga positiva en el interior de la célula por una falta de salida de sodio o por un aumento en la entrada de potasio. Se prolonga la repolarización ventricular y por tanto alarga el intervalo QT, predisponiendo a la TdP. La prevalencia es de 1/2000, siendo el síncope la primera manifestación clínica más común. Los pacientes sintomáticos comienzan sobre los 12 años de edad en un 50 % de los casos y a los 40 en un 90 %., Es uno de los causantes de la muerte súbita infantil; el 10 % de estas muertes presentan una mutación genética causante del SQTL. Se han identificado numerosos tipos de SQTL congénitos (el síndrome de Romano-Ward subtipos del QTL1-QTL15 y el síndrome de Jervell y Lange-Nielson subtipos JLN1-JLN2), pero los más comunes son los incluidos en el síndrome de Romano-Ward, y entre ellos QTL1-3., Existen factores que predisponen a los eventos cardiacos según los distintos tipos de QTL; por ejemplo, el QTL-1 se estimula con la actividad física o el estrés emocional (natación), en el caso de QTL-2 por estímulos auditivos (como un despertador o un timbre), el postparto o el estrés emocional. Recientemente se ha observado que la epilepsia es más común entre el tipo 2, en un 39 %. Es importante el diagnóstico diferencial porque muchos se tratan erróneamente como epilepsia por las convulsiones que se dan durante la TdP., El diagnóstico del SQTL congénito se basa en el score de Schwartz (tabla 1), que valora hallazgos electrocardiográficos, historial clínico y familiar. Se diagnostica de SQTL congénito con un sumatorio de 3,5 o más y sin causas adquiridas. Existen casos de puntuación de 1 a 3 en los que la prolongación del QT está latente, pero se puede desenmascarar realizando un Holter o series electrocardiográficas durante el ejercicio físico o test de epinefrina3,4.,úngicos, antidepresivos tricíclicos, neurolépticos y procinéticos), interacción medicamentosa, periodos largos de ayuno o dietas líquidas proteicas, inhibidores enzimáticos (ingesta de zumo de pomelo), bradicardia sinusal o pausas por bloqueo sinoauricular o auriculoventricular, insuficiencia hepática o renal, cardiopatía estructural (cardiopatía isquémica, insuficiencia cardiaca, hipertrofia ventricular), sexo femenino (por poseer un intervalo QT más prolongado que el hombre en condiciones fisiológicas), edad avanzada, cardioversión por fibrilación auricular y accidente cerebrovascular agudo5.,
Para diagnosticar un SQTL, primero debemos saber medir correctamente el intervalo QT en el ECG. El intervalo QT comprende desde el inicio del complejo QRS hasta el principio de la onda T, y se debe medir donde se pueda visualizar la onda Q (normalmente en DII, V5 o V6). La medición del QT debe ajustarse a la frecuencia cardiaca; a eso se le llama QT corregido (QTc) y se calcula con la fórmula de Bazett.
QTc= QT/√RR
El intervalo RR es la distancia que hay entre la onda R y la siguiente onda R., Ambos valores se deben medir en milímetros (mm) y pasarlos a segundos antes de introducirlos en la fórmula. Se miden los mm y se multiplica por 0,04. En caso de arritmia se deben hacer varias mediciones y se calcula la media. Debemos poner especial atención a no incluir la onda U en la onda T. En caso de no poder diferenciarse bien el final de la onda T se aconseja usar el método de la tangente, que consiste en considerar que la onda T acaba en la intersección de la tangente de la porción más inclinada de la descendente de la onda T y la línea de base (figura 3)., Los valores normales del intervalo QT se distinguen entre menores de 12 años y mayores de 12 años. En los mayores de 12 años se diferencia entre varones y mujeres, considerándose normal entre 0,39 y 0,45 en los primeros y entre 0,39 y 0,46 en las segundas3.

La prolongación del intervalo QT puede predisponer a una torsión de puntas o TdP. La TdP es una variación polimorfa irregular del QRS que va cambiando de positivo a negativo para volver a empezar; parece que las puntas se torsionan alrededor del eje eléctrico., La frecuencia cardiaca es de entre 150 y 300 lpm con intervalos RR irregulares. Comienza de forma súbita y no es sostenida.
El tratamiento de la TdP es cardioversión en caso de que haya compromiso hemodinámico. Se deben retirar todos los fármacos que puedan producir el alargamiento del QT. El sulfato de magnesio es de primera elección ya sea congénito o adquirido e independiente del nivel de magnesio en suero: un bolo de 2 g a pasar en 2-3 minutos seguido de infusión iv 2-4 mg/min y pudiendo pasar un segundo bolo si la TdP se repite mientras se está poniendo la infusión iv., Es compatible con el embarazo. Puede producir hipertensión arterial y asistolia. El antídoto del sulfato de magnesio es el gluconato cálcico. El isoproterenol se usa cuando fracasa el sulfato de magnesio y el marcapasos, o el personal entrenado no está entrenado para colocar el marcapasos. No se debe usar en el SQTL congénito. Como efectos secundarios son comunes las palpitaciones, enrojecimiento facial y calor. En cuanto a los niveles de potasio séricos, se ha demostrado que es preferible que estén en los límites altos de la normalidad, pudiendo requerir su administración iv., No está contraindicado en el embarazo y nunca debe administrarse en bolo porque puede producir arritmias y depresión cardiaca provocando la muerte. Se debe monitorizar con ECG. El marcapasos temporal endovenoso se puede colocar a unos 100 lpm si el magnesio no logra prevenir la TdP.
El tratamiento a largo plazo del SQTL en caso de ser adquirido no es necesario, porque una vez que se elimina la causa suele ceder. En cambio, si es congénito es aconsejable el uso de β-bloqueantes por vía oral al disminuir de un 0,97 a un 0,31 los eventos cardiacos por paciente y año., Se aconseja mantener una frecuencia cardiaca durante el ejercicio inferior a 130 lpm. Los suplementos de potasio son recomendables en QTL-2. El desfibrilador implantable y la denervación simpática cardiaca izquierda se recomiendan en pacientes con antecedentes de parada cardiaca, clínica antes del año de vida o JLN-1. La educación es importante para evitar los estímulos precipitantes (ejercicio físico, natación, falta de sueño, estímulos auditivos, estímulos simpáticos intensos, dolor, sufrimiento, enfado sobresaltos)3,6-8.,
Nos parece que la medición del QTc es algo pasado por alto en atención primaria a pesar de ser fácil de calcular y muy accesible, puesto que el ECG es una prueba complementaria muy común en nuestra labor diaria. El simple hecho de reconocerlo y actuar en consecuencia puede prevenir graves consecuencias. La medición sistemática del intervalo QT para su detección precoz en tratamiento con medicación que lo alarga podría ser recomendable, al igual que en las alteraciones electrolíticas.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.,
Bibliografía
1. Kallergis EM, Goudis CA, Simantirakis EN, Kochiadakis GE, Vardas PE. Mechanisms, risk factors, and management of acquired long QT Syndrome: a comprehensive review. Scientific World Journal. 2012; 2012: 212178.
3. Muñoz Castellano J. Síndrome de QT largo y Torsade de Pointes. Emergencias. 2004; 16: 85-92.
















Deja una respuesta