Male Turner syndrome, Noonan-Ehmke syndrome, Turner-like syndrome, Ullrich-Noonan syndrome
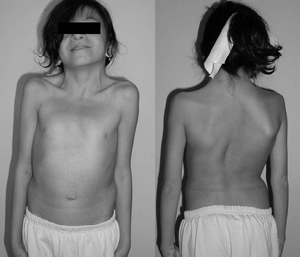
una niña de 12 años con síndrome de Noonan. Típico cuello palmeado. Doble curva estructural con deformidad costal.,irmed con pruebas genéticas
síndrome Cardiofaciocutáneo, síndrome de Turner, síndrome de Costello, neurofibromatosis tipo 1
basado en los síntomas
hormona de crecimiento
depende de la gravedad de los problemas cardíacos
1 de cada 100 (1 de cada 2.000 enfermedades graves)
El síndrome de Noonan (SN) es un trastorno genético que puede presentarse con rasgos faciales ligeramente inusuales, estatura baja, enfermedad cardíaca congénita, problemas de sangrado y malformaciones esqueléticas., Las características faciales incluyen ojos ampliamente espaciados, ojos de color claro, orejas bajas, un cuello corto y una mandíbula inferior pequeña. Los problemas cardíacos pueden incluir estenosis de la válvula pulmonar. El esternón puede sobresalir o estar hundido, mientras que la columna vertebral puede estar anormalmente curvada. La inteligencia en el síndrome suele ser normal. Las complicaciones del SN pueden incluir leucemia.
una serie de mutaciones genéticas pueden resultar en el síndrome de Noonan. La afección puede heredarse de los padres de una persona como una afección autosómica dominante o presentarse como una nueva mutación., El síndrome de Noonan es un tipo de Rasopatía, cuyo mecanismo subyacente implica la sobreactivación dentro de la vía de señalización celular RAS/MAPK. El diagnóstico se puede sospechar sobre la base de los síntomas, las imágenes médicas y los análisis de sangre. La confirmación se puede lograr con pruebas genéticas.
no se conoce cura para el NS. El tratamiento se basa en los síntomas y los problemas subyacentes, y es posible que se requiera apoyo adicional en la escuela. La terapia con hormona del crecimiento durante la infancia puede aumentar la altura final de una persona afectada. Los resultados a largo plazo generalmente dependen de la gravedad de los problemas cardíacos.,
se estima que 1 de cada 1.000 personas se ven levemente afectadas por el SN, mientras que aproximadamente 1 de cada 2.000 tienen una forma más grave de la afección. Los hombres parecen ser afectados con más frecuencia que las mujeres. La afección se describió por primera vez en 1883 y fue nombrada en honor a la cardióloga pediátrica estadounidense Jacqueline Noonan, quien describió más casos en 1963.
signos y síntomas

características anormales del síndrome de Noonan a la edad de 3 meses: observe la inclinación de la ceja y la caída del párpado izquierdo.,

características Anormales del síndrome de Noonan, a la edad de 3 meses: Nota la baja, hacia atrás, girar, y de forma anormal en el oído.
los signos más comunes que conducen al diagnóstico del síndrome de Noonan son las características faciales únicas y las características musculoesqueléticas. Las características faciales son más prominentes en la infancia, haciéndose menos evidentes con la edad en muchas personas con síndrome de Noonan.,
cabeza
algunas de las características del síndrome de Noonan incluyen una cabeza grande con exceso de piel en la parte posterior del cuello, línea baja del cabello en la nuca, línea alta del cabello en la parte delantera de la cabeza, forma triangular de la cara, frente ancha y un cuello corto y palmeado.
en los ojos, el hipertelorismo (ojos ampliamente establecidos) es una característica definitoria, presente en el 95% de las personas con síndrome de Noonan., Esto puede ir acompañado de pliegues epicantales (pliegue adicional de piel en la esquina interna del ojo), ptosis (caída de los párpados), proptosis (ojos saltones), estrabismo (giro hacia adentro o hacia afuera de los ojos), nistagmo (movimiento brusco de los ojos) y errores visuales refractivos.
la nariz puede ser pequeña, ancha y hacia arriba.
el desarrollo de la boca también puede verse afectado en el síndrome de Noonan., Esto puede resultar en un surco profundo del filtro (línea del labio superior) (más del 90%), micrognatia (mandíbula inferior de tamaño insuficiente), paladar arqueado alto, dificultades de articulación (los dientes no se alinean) que pueden conducir a problemas dentales. Similar a las manifestaciones musculares anteriores, en la boca, se puede observar un mal control de la lengua.,
piel
los signos y síntomas cutáneos en el síndrome de Noonan incluyen linfedema (hinchazón linfática de las extremidades), formación de queloides, formación excesiva de cicatrices, hiperqueratosis (sobredesarrollo de la capa externa de la piel), nevos pigmentados (manchas cutáneas de pigmentación oscura) y enfermedad del tejido conjuntivo
en el síndrome de Noonan pueden producirse anomalías musculoesqueléticas
en las extremidades. Esto puede manifestarse como dedos terminados sin rodeos, relleno adicional en los dedos de las manos y los pies, edema del dorso de las manos y la parte superior de los pies, y cúbitus valgus (ángulo de transporte amplio de los codos).,
para la baja estatura, la hormona del crecimiento a veces se combina con IGF-1 (o como alternativa, IGF-1 como independiente) se puede utilizar para lograr una mayor altura/altura final más rápido. La estatura adulta final de los individuos con síndrome de Noonan es de aproximadamente 161-167 cm en los hombres y 150-155 cm en las mujeres, que se acerca al límite inferior de la normalidad.
las anomalías espinales pueden estar presentes hasta el 30% de las veces y esto puede requerir cirugía para corregir en más del 60% de estos casos., Otras manifestaciones musculoesqueléticas en el síndrome de Noonan están asociadas con trastornos indiferenciados del tejido conjuntivo que pueden estar asociados con contracturas articulares (opresión) o hipermovilidad articular (flojedad). Los factores adicionales pueden presentarse en forma de aleteo de la escápula, escoliosis, prominencia del esternón (pectus carinatum), depresión del esternón (pectus excavatum). Las anomalías musculares pueden presentarse como hipotonía (tono muscular bajo), que puede conducir a lordosis (aumento de hueco en la espalda) debido a un tono muscular abdominal deficiente.,
corazón
el síndrome de Noonan es la segunda causa sindrómica más común de cardiopatía congénita. Esto incluye estenosis valvular pulmonar (50-60%), comunicación interauricular (10-25%), comunicación interventricular (5-20%) y miocardiopatía hipertrófica (12-35%).
pulmones
En algunas personas se ha notificado una función pulmonar restrictiva.
Gastrointestinal
varios síntomas gastrointestinales (GI) se han asociado con el síndrome de Noonan., Estos incluyen dificultades para tragar, baja motilidad intestinal, gastroparesia (retraso en el vaciado gástrico), malrotación intestinal y vómitos frecuentes o fuertes. Estos problemas digestivos pueden conducir a una disminución del apetito, retraso en el desarrollo desde la infancia hasta la pubertad (75%) y, en ocasiones, la necesidad de una sonda de alimentación.{ Se ha asociado con la enfermedad de Von Willebrand, trombocitopenia Amegacariocítica (bajo recuento de plaquetas), tiempo prolongado de tromboplastina parcial activada, defectos de coagulación combinados., Cuando están presentes, estos trastornos acompañantes del síndrome de Noonan pueden asociarse con una predisposición a contusiones fácilmente o hemorragia.
neurológica
ocasionalmente,puede ocurrir malformación de Chiari (tipo 1), que puede conducir a hidrocefalia. También se han notificado convulsiones.
Causas

NS normalmente se hereda en un patrón autosómico dominante con expresión variable.,
la recurrencia en hermanos y la transmisión aparente de padres a hijos han sugerido durante mucho tiempo un defecto genético con herencia autosómica dominante y expresión variable. Se sabe que las mutaciones en las vías de señalización de la proteína quinasa activada por Ras/mitógeno son responsables de aproximadamente el 70% de los casos de NS.
Las personas con NS tienen hasta un 50% de probabilidad de transmitirlo a su descendencia., El hecho de que un padre afectado no siempre se identifique para niños con NS sugiere varias posibilidades:
- Las manifestaciones pueden ser tan sutiles que no se reconocen (expresividad variable)
- ns es heterogénea, comprende más de una condición similar de diferentes causas, y algunas de estas pueden no ser hereditarias.
- Una alta proporción de casos puede representar mutaciones nuevas y esporádicas.,
| Type | Online Mendelian Inheritance in Man database | Gen | Year found | Locus | % of cases | Description | árbitros. |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | PTPN11 | 2001 | 12q24.1 | 50% | El gen PTPN11 codifica para la proteína tirosina fosfatasa SHP-2., Esta proteína es un componente de varias vías de transducción de señales intracelulares involucradas en el desarrollo embrionario que modulan la división celular, la diferenciación y la migración, incluida una mediada por el receptor del factor de crecimiento epidérmico, que es importante en la formación de las válvulas cardíacas semilunares.la duplicación de la región cromosómica que contiene PTPN11 también puede resultar en NS., | |
| NS2 | 605275 | Unknown; autosomal recessive | |||||
| NS3 | 609942 | KRAS | 2006 | 12p12.1 | <5% | ||
| NS4 | 610733 | SOS1 | 2006 | 2p21 | 10% | Activating mutations in SOS1 can give rise to NS. SHP-2 and SOS1 positively regulate the Ras/MAP kinase pathway, suggesting that its dysregulation mediates NS development., | |
| NS5 | 611553 | RAF1 | 2007 | 3p25 | 3-17% |
las mutaciones Heterocigotas en la ANR, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, y CBL también se han asociado con un menor porcentaje de NS y fenotipos relacionados.
una condición conocida como «neurofibromatosis-síndrome de Noonan» se asocia con neurofibromina.
diagnóstico
El NS puede confirmarse genéticamente por la presencia de cualquiera de las mutaciones conocidas enumeradas anteriormente., Sin embargo, a pesar de la identificación de 14 genes causantes, la ausencia de una mutación conocida no excluirá el diagnóstico, ya que más genes aún no descubiertos pueden causar NS. Por lo tanto, el diagnóstico de SN todavía se basa en las características clínicas. En otras palabras, se hace cuando un médico siente que una persona tiene suficientes características para justificar la etiqueta. Los valores principales de hacer un diagnóstico genético son que guía evaluaciones médicas y de desarrollo adicionales, excluye otras explicaciones posibles para las características y permite estimaciones de riesgo de recurrencia más precisas., Con más estudios de correlación genotipo-fenotipo que se realizan, un diagnóstico genético positivo ayudará al médico a ser consciente de posibles anomalías específicas de esa mutación genética determinada. Por ejemplo, se observa un aumento en la miocardiopatía hipertrófica en personas con una mutación de KRAS y existe un mayor riesgo de leucemia mielomonocítica juvenil para una mutación de PTPN11. En el futuro, los estudios pueden conducir a un manejo dirigido de los síntomas de NS que depende de la mutación genética que tenga una persona.,
antes del nacimiento
las características prenatales que podrían llevar a los médicos a considerar un diagnóstico de síndrome de Noonan incluyen higroma quístico, aumento de la translucencia nucal, derrame pleural y edema.
diagnóstico diferencial
Si bien el síndrome de Turner tiene similitudes con las anomalías renales y el retraso en el desarrollo, el síndrome de Turner solo se encuentra en mujeres y, a menudo, se expresa de manera diferente. En el síndrome de Turner, hay una menor incidencia de retrasos en el desarrollo, los defectos cardíacos del lado izquierdo son constantes y la aparición de anomalías renales es mucho menor.,
otras Rasopatías
- Síndrome de Watson-el síndrome de Watson tiene una serie de características similares con el síndrome de Noonan, como baja estatura, estenosis de la válvula pulmonar, desarrollo intelectual variable y cambios en la pigmentación de la piel.
- Síndrome Cardiofaciocutáneo (CFC) – el síndrome CFC es muy similar al síndrome de Noonan debido a características cardíacas y linfáticas similares. Sin embargo, en el síndrome de CFC la discapacidad intelectual y los problemas gastrointestinales Son a menudo más graves y pronunciados.,
- Síndrome de Costello-como el síndrome CFC, el síndrome de Costello tiene características superpuestas con el síndrome de Noonan. Sin embargo, las condiciones se pueden distinguir por su causa genética.
- Neurofibromatosis 1 (NF1)
- Síndrome de Williams
manejo
El tratamiento varía dependiendo de las complicaciones pero tiende a ser bastante estándar, reflejando el tratamiento de la población general., Las directrices de manejo, divididas por sistemas, incluyendo general, de desarrollo, dental, crecimiento y alimentación, cardiovascular, audiológico, hematológico, renal y esquelético, que tienen en cuenta las acciones que deben tomarse en el momento del diagnóstico, después del diagnóstico y si son sintomáticas, han sido publicadas por un consorcio estadounidense.
específicamente, el tratamiento de las complicaciones cardiovasculares se asemeja al de la población general y el tratamiento de la diátesis hemorrágica se guía por la deficiencia del factor específico o agregación plaquetaria.,
- Las pruebas neuropsicológicas se recomiendan para encontrar fortalezas y desafíos para adaptar el apoyo necesario para la escuela y la carrera.
- La personalización educativa, como un plan de programa de Educación Individualizado, a veces es necesaria para los niños en edad escolar.
- terapia del habla si los problemas del habla y la articulación presentan
- La fisioterapia y la terapia ocupacional para los retrasos motores gruesos y finos
- La hipotonía y las dificultades motoras a menudo afectan la escritura a mano. Las adaptaciones para disminuir las demandas de escritura a mano mejorarán el rendimiento y ahorrarán la función de la mano a largo plazo.,
- Se recomienda el seguimiento periódico y el control de por vida de las anomalías encontradas en cualquier sistema, especialmente el sistema cardiovascular.
riesgo de anestesia
aunque se ha informado que algunas personas con síndrome de Noonan desarrollan hipertermia maligna, la mutación genética de enfermedades que se sabe que están asociadas con la hipertermia maligna es diferente a la del síndrome de Noonan.,
pronóstico
la esperanza de vida de las personas con síndrome de Noonan puede ser similar a la de la población general, sin embargo, el síndrome de Noonan puede estar asociado con varias condiciones de salud que pueden contribuir a la mortalidad. El mayor contribuyente a la mortalidad en individuos con síndrome de Noonan son las complicaciones de la enfermedad cardiovascular. Por lo tanto, el pronóstico depende en gran medida de la presencia o ausencia de enfermedad cardíaca, así como del tipo y la gravedad de la enfermedad (si la enfermedad está presente)., En particular, el síndrome de Noonan con miocardiopatía hipertrófica se asocia con un aumento de la mortalidad.
historia
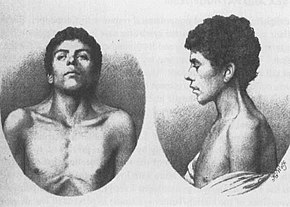
el caso más antiguo conocido de NS, descrito en 1883 por Kobylinski
Jacqueline Noonan ejercía como cardióloga pediátrica en el universidad de Iowa cuando notó que los niños con un tipo raro de defecto cardíaco, estenosis valvular pulmonar, a menudo tenían una apariencia física característica, con baja estatura, cuello palmeado, ojos espaciados y orejas bajas., Tanto los niños como las niñas se vieron afectados. Estas características se observaron a veces en familias, pero no se asociaron con anomalías cromosómicas macroscópicas. Estudió a 833 personas con síndrome de Noonan en la clínica de cardiopatías congénitas, buscando otras anomalías congénitas, y en 1963 presentó un artículo: «malformaciones No cardiacas asociadas en niños con cardiopatías congénitas». Se describieron 9 niños que además de cardiopatía congénita tenían rasgos faciales característicos, deformidades torácicas y baja estatura.
Dr. John Opitz, un ex alumno del Dr., Noonan, primero comenzó a llamar a la condición «síndrome de Noonan» cuando vio niños que se parecían a los que el Dr. Noonan había descrito. La Dra. Noonan produjo un artículo titulado «hipertelorismo con fenotipo de Turner» en 1968 donde estudió a 19 pacientes que mostraron síntomas indicativos del síndrome de Noonan. En 1971 en el Simposio de defectos cardiovasculares, el nombre’ síndrome de Noonan ‘ se reconoció oficialmente.
















Deja una respuesta