Introducción
la discapacidad intelectual (ID) afecta a casi el 1-2% de la población y es el trastorno del neurodesarrollo (NDD) más común. Se ha encontrado que un número considerable de pacientes con DH tienen una causa genética (revisado en Bessa et al., 2012)., Las técnicas de análisis genómico utilizadas actualmente para la investigación de la etiología a menudo conducen a la identificación de variantes casi privadas muy raras, siendo la recolección de pacientes con alteraciones en el mismo gen un aspecto crucial de la definición de una nueva entidad clínica.
a principios de este año, se han descrito pacientes con mutaciones en el gen EBF3, que presentan un síndrome del neurodesarrollo que incluye DH, ataxia, hipotonía, dismorfismos Faciales leves y anomalías genitourinarias (OMIM 617330) (Chao et al., 2017; Harms et al., 2017; Sleven et al., 2017)., El gen EBF3 (Early B cell Factor 3) codifica un miembro de la familia de factores de transcripción de células B tempranas altamente conservadas, expresadas en niveles altos en el sistema nervioso en desarrollo (datos recuperados del Portal GTEx). EBF3 es un objetivo transcripcional de ARX, y se ha demostrado que está regulado por NeuroD y ARX (Friocourt y Parnavelas, 2011). ARX codifica un factor de transcripción crítico para el desarrollo embrionario que, durante muchos años, se ha asociado con una amplia gama de trastornos del neurodesarrollo., El deterioro intelectual, sistema nervioso central y anomalías genitourinarias observadas en pacientes con ambas mutaciones en EBF3 y ARX podrían reflejar la contribución de ambas proteínas a los mismos procesos moleculares y celulares (Chao et al., 2017). La función EBF3 también se ha estudiado en modelos animales. La ablación de sus ortólogos en gusanos y moscas conduce a un deterioro del desarrollo neuronal (Prasad et al., 1998; Hattori et al., 2013)., En ratones, la eliminación de Ebf3 conduce a la letalidad neonatal y defectos de migración neuronal, con fallas en el proyecto de neuronas olfativas en el bulbo olfativo dorsal (Wang et al., 2004). Los mecanismos patógenos exactos de las mutaciones EBF3 aún no están completamente dilucidados, pero el tipo de variantes descritas hasta ahora sugieren que la haploinsuficiencia, la ganancia de función y la negativa dominante son posibles mecanismos patógenos para las variantes descritas (Chao et al., 2017; Sleven et al., 2017).,
en este trabajo contribuimos con un paciente con la deleción más pequeña (600 Kb) notificada hasta la fecha que afecta a la totalidad del gen EBF3 y con una presentación clínica superpuesta a la de los pacientes con variantes de nucleótido único (SNVs) EBF3. Además, hacemos una comparación clínica de los pacientes con deleciones terminales grandes 10Q publicadas previamente y reportamos que, a pesar de las diferencias de tamaño, existe una superposición fenotípica significativa entre los pacientes con estas alteraciones., Estos hallazgos se suman al conocimiento actual de los trastornos relacionados con EBF3 y apoyan la HAPLOINSUFICIENCIA de EBF3 como clave en el síndrome de Neurodesarrollo asociado con las deleciones 10qter.
materiales y métodos
El paciente fue determinado dentro de un amplio estudio de trastornos del neurodesarrollo en Portugal, en el que la inscripción de los pacientes y las familias fue realizada por el médico remitente, la información clínica fue recopilada en una base de datos anónima de acuerdo con la autoridad portuguesa de protección de datos (CNPD) y se obtuvo el consentimiento informado por escrito de todos los participantes., El consentimiento informado para el presente paciente fue proporcionado por la madre para el estudio genético y la publicación de los Resultados (incluyendo fotos). El estudio fue aprobado por el Comité de ética del Centro de Genética Médica Dr. Jacinto Magalhães, Instituto Nacional de Salud Dr. Ricardo Jorge.
se extrajo ADN genómico de sangre periférica utilizando el kit de aislamiento de ADN Citogene® (Citomed, Portugal). aCGH se realizó utilizando Agilent 180 K array (AMADID:023363) contra una referencia de ADN diploide (Kreatech’s MegaPoll Reference DNA, Kreatech Diagnostics, Amsterdam)., aCGH analysis was performed using the Nexus Copy Number 6.0 software with FASST2 Segmentation algorithm (BioDiscovery Inc., El Segundo, CA). Genomic coordinates are according to Human Genome Build hg19. CNV confirmation was performed by qPCR for EBF3 (forward primer—CTCTCTGCTGGGTGCTGAG; reverse primer—GCGTCCCTTCATACGCTAAC; ENST00000368648.7) gene and using SDC4 (forward primer—ACCGAACCCAAGAAACTAGA; reverse primer—GTGCTGGACATTGACACCT; ENSG00000124145, Chr.20) and ZNF80 (forward primer—GCTACCGCCAGATTCACACT; reverse primer—AATCTTCATGTGCCGGGTTA; ENSG00000174255, Chr.3) as references genes., El análisis se realizó en una máquina de PCR en tiempo real 7500-FAST (Applied Biosystems, Foster City, CA, USA) utilizando Power SYBR Green® (Applied Biosystems, Foster City, CA, USA) de acuerdo con las recomendaciones del fabricante y siguiendo las directrices generales para qPCR. La especificidad de cada reacción se verificó mediante la generación de una curva de fusión para cada uno de los fragmentos amplificados. La eficiencia del cebador se calculó mediante la generación de una curva estándar que se ajustaba al porcentaje de eficiencia normal aceptado (cebadores utilizados enumerados en los datos complementarios)., Los valores de TC obtenidos para cada prueba fueron analizados en el software DataAssist™ (Applied Biosystems, Foster City, CA, USA).
resultados
aquí describimos un paciente con una deleción de novo que afecta a EBF3. Se trata de una niña de 11 años con DH grave (cociente de desarrollo global = 27 a los 7 años), nacida de padres no consanguíneos y sin antecedentes familiares de trastornos del neurodesarrollo. Nació después de un embarazo biamniótico bicoriónico gemelo (su hermana está sana), por parto vaginal, a las 35 semanas de gestación. Los parámetros de nacimiento fueron: Peso, 1.830 g (P3); longitud, 42.,5 cm (P10); y OFC, 30,6 cm (P10), con una puntuación de Apgar de 8/9 (1º y 5º min, respectivamente). El período neonatal se complicó con sepsis y el diagnóstico de esferocitosis hereditaria (heredada de su madre). Se observó un retraso global del desarrollo en los primeros meses, con control de la cabeza logrado a los 12 meses, sentado a los 18 meses, caminando independiente a los 30 meses y sin palabras pronunciadas a la edad de 3 años., Tenía pielonefritis a los 19 meses (la ecografía renal no mostró anomalías), reflujo gastroesofágico y otitis media recurrente, con pérdida auditiva conductiva que requirió intervención quirúrgica y un audífono. Se sospechó epilepsia a los 5 meses (episodios de actividad suspendida) pero el EEG fue normal.
se observó por primera vez a la edad de 3 años y 5 meses (Figuras 1A,B), momento en el que presentó hipotonía muscular, cara hipotónica, estrabismo y sensibilidad reducida al dolor., También presentó rasgos dismórficos leves (figura 1a): cara triangular, orejas pequeñas y bajas con anti-hélice prominente, cejas arqueadas, narinas antevertidas, punta nasal bulbosa, boca pequeña con esquinas hacia abajo, mentón puntiagudo, cuello corto y almohadillas fetales de dedo prominentes, así como una estatura baja leve (89 cm, correspondiente a alrededor de 2D).

la Figura 1. (A) aspecto Facial del paciente a los 3 años y 5 meses mostrando las orejas pequeñas y bajas con anti-hélice prominente y (B) almohadillas fetales en los dedos., C) aspecto Facial del paciente a los 11 años de edad. (D) resaltada en gris la deleción de 600 Kb en la región 10Q26.3; un zoom del gen EBF3 en la base de datos de la DGV revela la existencia de 3 deleciones en 3 controles que afectan a los primeros 6 exones de EBF3 (NM_001005463); los CNVs dentro de esta región encontrados en poblaciones de control incluyen deleciones nvs825626 (presente en 1/31 individuos), nvs552315 (presente en 1/17421 individuos) y nsv552316 (presente en 1/31 individuos). E) la representación esquemática de las transcripciones EBF3.,
la RM Cerebral se realizó a los 6 años, pero no se observaron anomalías. A la edad de 10 años fue reevaluada; todavía tenía otitis media recurrente, pero por lo demás estaba en buena salud global. El lenguaje era muy pobre (dos frases habladas después de 8 años). Tenía problemas de comportamiento, con movimientos estereotipados (movimientos rotativos, masticación de ropa, retropulsión de cabeza), puntuación para trastorno del espectro autista severo (ADI-R y ADOS) a los 7 años; mostró agitación y comportamiento agresivo (auto y hetero) y fue medicada con medicamentos antipsicóticos., Se realizó una cirugía ortopédica para el pes plano. Los rasgos faciales fueron similares a los descritos anteriormente, con incisivos centrales superiores espaciados (figura 1C); tenía anteojos para estrabismo e hipermetropia.
el análisis del ADN genómico por aCGH reveló una deleción de novo de 600 kb en 10p26.3 (Figura 1D) que afecta a tres genes: MGMT (que codifica la enzima o—6-metilguanina-ADN metiltransferasa, implicada en la reparación del ADN), EBF3 y GLRX (que codifica la glutaredoxina, una pequeña tioltransferasa que elimina los aductos de la proteína GSH), de los cuales EBF3 fue el gen más probable asociado a la enfermedad.,
discusión
el paciente presentado fue analizado por primera vez por la aCGH hace unos años. En el momento del análisis de aCGH, la existencia de las tres variantes presentes en la base de datos de variantes genómicas (DGV)1 que afectan a los cinco primeros exones del gen EBF3 (figura 1e; Park et al., 2010; Cooper et al., 2011), así como la ausencia de otras enfermedades conocidas causantes de mutaciones en este gen, nos llevan a clasificarlo como una variante de significación desconocida (VOUS). Sin embargo, las publicaciones recientes de mutaciones EBF3 (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) y las similitudes clínicas con los casos reportados, nos llevan a reevaluar la variante y nos hacen creer que la deleción de EBF3 puede ser de hecho responsable de la enfermedad en el paciente. Uno de los aspectos que plantearon dudas sobre la patogenicidad de esta variante en primer lugar fue la existencia de controles poblacionales con deleciones de los primeros seis exones de este gen, en heterocigosidad (datos extraídos de la base de datos de la DGV a febrero de 2017) (figura 1D). Aunque una transcripción de EBF3 que comienza en Exon13b está listada en la base de datos Ensembl (Enst00000440978.,1) (Figura 1e), que podría explicar cómo la deleción de los primeros exones podría eventualmente resultar en un fenotipo normal, esta transcripción excluye el dominio de unión al ADN de EBF3, y su patrón de expresión y relevancia funcional no han sido caracterizados. Sin embargo, tras la reevaluación, los CNV descritos por Cooper y sus colegas (nsv552315 y nsv552316) (Chao et al., 2017; Harms et al., 2017; Sleven et al.,, 2017) se consideraron en el umbral de detección por microarray SNP y no pueden ser la base para la exclusión de un gen candidato, particularmente a la luz de la fuerte evidencia genética y funcional de la relevancia de las mutaciones EBF3 para la enfermedad (Evan Eichler, Greg Cooper y Bradley Coe, comunicación personal).,
nuestro paciente muestra muchas similitudes clínicas con pacientes previamente descritos con mutaciones en EBF3 (21 casos resumidos en la tabla 1), como retraso global del desarrollo, retraso del habla expresiva, hipotonía, aumento del umbral de dolor, problemas de comportamiento y rasgos faciales característicos (cara larga/triangular, frente grande, Cara hipotónica)., Sin embargo, a pesar de que nuestro paciente tuvo un retraso significativo en el desarrollo motor, no se detectó ataxia significativa (con algunas limitaciones en el examen clínico, ya que el niño no cooperó), y no se presentaron anomalías cerebelosas en la RM cerebral.

la Tabla 1. Comparación clínica del presente caso con los casos reportados con mutaciones puntuales / indels en el gen EBF3.
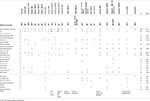
la Tabla 2., Comparación clínica del presente caso con los casos reportados con deleciones que afectan al citoband 10q26.
las variantes EBF3 descritas en la literatura a principios de 2017 incluyen mutaciones puntuales que se predice que son deletéreas y pequeñas inserciones y eliminaciones que conducen a la deleción en el marco de aminoácidos clave o a un cambio de Marco, causando predeciblemente el truncamiento temprano de la proteína resultante o la descomposición mediada por tonterías., Las mutaciones se concentraron en partes del gen que codifica el dominio de unión al ADN de EBF3, y se predijeron a través de diferentes métodos para conducir a una pérdida de función de este factor de transcripción, lo que sugiere una función reducida y haploinsuficiencia como el mecanismo subyacente a la alteración del neurodesarrollo en estos pacientes. Se describe que los ratones Knock-out para Ebf3 presentan letalidad neonatal y defectos de migración neuronal, con falla de las neuronas olfativas para proyectarse al bulbo olfativo dorsal (Wang et al.,, 2004), pero no se hace ninguna descripción de un fenotipo en los animales heterocigotos, que en realidad se presentan como controles en muchos de los experimentos, por lo que no se apoya el modelo de haploinsuficiencia. Hicimos esfuerzos para obtener y estudiar el fenotipo del neurodesarrollo de estos animales, pero no tuvimos éxito, ya que la línea de knock-out Ebf3(o/E2) puede haber sido descontinuada (Joseph W. Lewcock, comunicación personal). Sin embargo, el caso actual, junto con los pacientes resumidos en la Tabla 2, sí apoyan la hipótesis de la HAPLOINSUFICIENCIA del FEB3 como causante de la enfermedad.,
En resumen, la descripción Actual refuerza la pérdida de función / haploinsuficiencia del EBF3 como causa de enfermedad del neurodesarrollo, y refuerza la Asociación de este gen con un síndrome clínico característico dentro de este espectro.
contribuciones de los autores
FL realizó los estudios moleculares y analizó los datos moleculares. MG y GS recogieron datos clínicos. FL, JP y PM revisaron todos los casos de mutación EBF3 en la literatura. FL, GS, JP y PM redactaron el documento. PM obtuvo financiación para este estudio. El estudio fue realizado bajo la dirección de PM.,
financiación
FCT—Fundação para a Ciência e a Tecnologia dentro de los proyectos y becas (PIC/IC/83026/2007, PIC/IC/83013/2007, SFRH/BD/90167/2012). Este artículo ha sido desarrollado en el marco del proyecto NORTE-01-0145-FEDER-000013, apoyado por el Programa Operativo Regional Norte de Portugal (NORTE 2020), en el marco del Acuerdo de Asociación Portugal 2020, a través del Fondo Europeo de Desarrollo Regional (FEDER).
Declaración de conflicto de intereses
JP fue empleado por la compañía CGC Genetics.,
los demás autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran ser interpretadas como un potencial conflicto de intereses.
agradecimientos
queremos agradecer a la paciente y a su familia por su participación en los estudios genéticos y por permitir esta publicación. Agradecemos a la Dra. Ana María Fortuna y a la Dra. Margarida Reis Lima por hacer posible este proyecto y por facilitar nuestra colaboración con CGM.
notas al pie
1., ^Base de datos de variantes genómicas disponible en: http://dgv.tcag.ca/dgv/app/home.
Friocourt, G., and Parnavelas, J. (2011). La identificación de objetivos Arx revela nuevos candidatos para controlar la migración y diferenciación de interneuronas corticales. Delantero. Celular. Neurociencia. 5:28. doi: 10.3389 / fncel.2011.00028
resumen de PubMed / texto completo de CrossRef / Google Scholar
















Deja una respuesta