US Pharm. 2010;35(5):36-41.
una relación entre la diabetes mellitus (DM) y la demencia es innegable, con numerosos estudios que concluyen que la DM aumenta el riesgo de deterioro cognitivo y demencia, incluida la enfermedad de Alzheimer (EA).1-5 la DM no solo aumenta el riesgo de demencia, sino que en realidad aumenta la tasa de desarrollo de demencia de dos a tres veces.,3 el mecanismo de este deterioro no se comprende completamente, pero se plantea la hipótesis de que la hiperglucemia, la resistencia a la insulina, el estrés oxidativo, los productos finales de la glicación avanzada y las citocinas inflamatorias conducen colectivamente a la disfunción cognitiva.5 de hecho, la diabetes fue descrita como un «tipo especial de envejecimiento acelerado» en 1976 debido a sus muchas complicaciones asociadas.,5 la aparente superposición entre DM y demencia ha llevado a la sugerencia de que la EA no es solo un trastorno neurológico, sino más bien un trastorno neuroendocrino, con Steen et al acuñando el término diabetes tipo 3 para describir esta enfermedad híbrida.,6
resumen de la resistencia a la insulina/alteración de la tolerancia a la glucosa
Fisiopatología: tanto la alteración de la tolerancia a la glucosa—definida como una glucosa plasmática de 140 mg/dL a 199 mg/dL después de una prueba de tolerancia oral a la glucosa—como la alteración de la glucosa en ayunas—definida como una glucosa plasmática en ayunas de 100 mg/dL a 125 mg/dL—son diagnósticos de prediabetes (precursora de la DM tipo 2 ), Al igual que una hemoglobina A1C de 5,7% a 6,4%.7 la fisiopatología de la prediabetes es la misma que la de la DMT2, que se diagnostica después de una glucemia en ayunas de 126 mg/dL o superior o una A1C de 6.,5% o más (prueba repetida para confirmación).7 en ambas condiciones, la resistencia a la insulina se desarrolla en los tejidos periféricos, donde se requiere insulina para la absorción de glucosa (es decir, músculo, hígado y grasa), y el páncreas se ve obligado a suministrar cada vez más insulina para superar esta resistencia.8 en esta etapa, los pacientes a menudo son hiperinsulinémicos; sin embargo, con el tiempo, el páncreas no puede satisfacer las demandas de los tejidos, y las células beta productoras de insulina del páncreas fallan lentamente, produciendo cada vez menos insulina.,8 sin suficiente insulina, las concentraciones de glucosa en sangre aumentan, dando lugar a prediabetes y, hasta en el 70% de los pacientes con prediabetes, A dm2.9 Las complicaciones de la hiperglucemia son muchas y abarcan un mayor riesgo de complicaciones macrovasculares (como infarto de miocardio, accidente cerebrovascular y enfermedad vascular periférica) y complicaciones microvasculares (como nefropatía, neuropatía y retinopatía).7
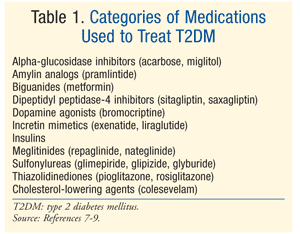
Tratamiento: existen muchas terapias para tratar la DM., El tratamiento inicial para la prediabetes y la T2DM es la modificación del estilo de vida que implica dieta, ejercicio y pérdida de peso.7 la American Diabetes Association (ADA) también recomienda la consideración de la metformina en pacientes con prediabetes que están en alto riesgo de desarrollar diabetes.7 se recomienda la metformina como tratamiento farmacológico inicial para la DMT2.7 a menudo, se necesita terapia farmacológica adicional para alcanzar los objetivos glucémicos de ADA, incluyendo un A1C <7%.,7 existen numerosas clases de medicamentos para la DMT2 (tabla 1), y la elección del agente para normalizar la glucosa en sangre depende de una serie de factores específicos del paciente y del medicamento. Es importante tener en cuenta que muchos pacientes con DMT2 eventualmente requerirán insulina exógena para lograr y mantener la euglucemia a medida que la función de las células beta disminuye progresivamente.
Descripción general de la EA
Fisiopatología: la EA es la forma más común de demencia en adultos mayores, representando del 60% al 80% de todos los casos.,10 dos características histopatológicas de la EA son las placas neuríticas y los enredos neurofibrilares.11 Las placas consisten en proteína beta-amiloide insoluble. Los enredos, que son intracelulares, están compuestos de proteína tau fosforilada. La proteína Tau es importante para el ensamblaje de microtúbulos, y cuando esta proteína está anormalmente fosforilada, la función neuronal se interrumpe. Los enredos neurofibrilares afectan más comúnmente a las neuronas colinérgicas.12 además, la interrupción del principal neurotransmisor excitador, el glutamato, contribuye a la patología de la EA., En la EA, hay una sobreactivación del glutamato, lo que indica un proceso conocido como excitotoxicidad, que conduce a la formación de placa, hiperfosforilación de la proteína tau y muerte celular.12 En resumen, la excitotoxicidad y la formación de placas neuríticas y enredos neurofibrilares interrumpen las vías de los neurotransmisores, lo que resulta en el deterioro del aprendizaje y la memoria asociado con la EA.
Tratamiento: las terapias farmacológicas aprobadas por la FDA disponibles para la EA se resumen en la tabla 2.,11,12 si bien todavía queda mucho por aprender con respecto a las terapias que tratan o alteran la patología de la EA, actualmente las terapias disponibles implican el uso de inhibidores de la acetilcolinesterasa, de los cuales hay cuatro, y el antagonista del receptor de N-metil-D-aspartato memantina.

La insulina y el cerebro
aunque es necesaria para el transporte de glucosa a los tejidos periféricos, la insulina no parece ser necesaria para el transporte de glucosa al cerebro o para el metabolismo cerebral de la glucosa., Mientras que el transporte de glucosa al cerebro no depende de la insulina, la insulina en sí es transportada a través de la barrera hematoencefálica (BBB) por los procesos de transporte mediados por el receptor de insulina.13 Este mecanismo de transporte es saturable; durante períodos prolongados de concentraciones excesivas de insulina (hiperinsulinemia) en la periferia, como los observados en prediabetes y T2DM, este mecanismo de transporte mediado por receptores se regula a la baja, reduciendo así el transporte de insulina hacia el cerebro y el líquido cefalorraquídeo.,
además de la insulina, el factor de crecimiento similar a la insulina tipo 1 (IGF-1) está presente en el cerebro y es necesario para el crecimiento y la función normales del sistema nervioso central (SNC).14 los receptores de insulina e IGF-1 están ubicados en todo el cerebro en neuronas y astrocitos (células en forma de estrella en el SNC que ayudan a apoyar a las neuronas en el cerebro). Dos áreas del cerebro que son cruciales para el aprendizaje y la memoria, el hipocampo y el hipotálamo, contienen altas concentraciones de receptores de insulina.5 en ratas, se ha demostrado que la administración intracerebroventricular de insulina mejora la memoria.,15 además, se ha encontrado que la administración intranasal de insulina en humanos aumenta el rendimiento de la memoria.16 Como se señaló anteriormente, la insulina no es necesaria para el metabolismo cerebral de la glucosa; sin embargo, puede haber áreas específicas en el cerebro en las que la insulina desencadena procesos metabólicos que involucran a la glucosa. En estudios en ratas, se ha demostrado que la insulina influye en la utilización de la glucosa en el hipotálamo y el locus coeruleus, dos áreas que son importantes para el aprendizaje y la memoria.,17 También se ha demostrado que el aprendizaje facilita una mayor expresión de los receptores de insulina del SNC, lo que lleva a la postulación de que la presencia y la actividad de la insulina en el cerebro contribuyen y desempeñan un papel esencial en el aprendizaje y la memoria. La insulina también puede contribuir a la modulación de los neurotransmisores del SNC, en particular la acetilcolina y la norepinefrina, los cuales son críticos para la cognición normal y la salud del cerebro.,18
resistencia a la insulina y demencia
Descripción general: parece haber muchos defectos en la señalización de la insulina en los cerebros de los pacientes con EA, lo que lleva a una disminución de la utilización de glucosa y el metabolismo energético.6,19 como la DMT2 se asocia con la resistencia a la insulina periférica, la EA se asocia con la resistencia a la insulina cerebral.20 al comienzo de la EA, parece haber una absorción inadecuada de insulina y señalización en el cerebro, un signo de resistencia a la insulina.5,19,21,22 se ha observado un aumento de los receptores de insulina en el cerebro de pacientes con EA, probablemente como compensación de la resistencia a la insulina.,21
En general, este deterioro en la señalización de la insulina tiene muchos efectos posteriores en la EA. Una teoría es que las bajas concentraciones de insulina en el SNC causan una disminución en los niveles de acetilcolina y el flujo sanguíneo cerebral.Además, las alteraciones en las concentraciones de insulina pueden promover la formación de proteínas beta-amiloide y tau.2 en el cerebro, la enzima degradante de la insulina (IDE) participa en la degradación y el aclaramiento de las proteínas beta-amiloides.Los niveles altos de insulina inhiben el IDE y pueden conducir a una disminución en el aclaramiento beta-amiloide, aumentando posteriormente la deposición cerebral de beta-amiloide.,5,19,21
recientemente las ceramidas, una familia de lípidos, han llamado la atención por su papel potencial en la resistencia a la insulina y la demencia.23,24 ceramidas se generan en presencia de inflamación, que es común en la obesidad, T2DM, y EA.24 debido a que la ceramida cruza fácilmente la BBB, la exposición causa un deterioro del metabolismo energético y alteraciones en la expresión génica de la insulina, contribuyendo a la resistencia a la insulina.23,24 esta es actualmente un área activa de interés, con investigadores que demuestran que la inhibición de la síntesis de ceramida previno la resistencia a la insulina mediada por la obesidad.,24
insulina Intranasal: como se mencionó anteriormente, la administración de insulina intranasal mejora el rendimiento de la memoria en humanos.16 se han realizado varios estudios de insulina intranasal. Tras la administración de 20 unidades de insulina intranasal dos veces al día a sujetos humanos, Reger et al encontraron que, en comparación con el placebo, el grupo tratado con insulina retuvo más información verbal y mostró una mejor atención y estado funcional.,25 Estos investigadores también notaron que la glucosa plasmática en ayunas y la insulina no cambiaron con el uso de insulina intranasal, lo que indica que la administración intranasal proporciona acceso directo a la insulina en el SNC sin efectos periféricos.Agonistas PPAR-Gamma: ampliamente estudiados para el tratamiento de la resistencia periférica a la insulina, las tiazolidindionas rosiglitazona y pioglitazona están siendo investigadas por su papel en el tratamiento de la EA. Las tiazolidindionas son agonistas gamma-receptores activados por proliferadores de peroxisomas (PPAR)., PPAR-gamma está involucrado en el metabolismo de la glucosa y los lípidos y modula la inflamación, un contribuyente conocido a la EA.22,27 las Tiazolidindionas son sensibilizadores de insulina, que trabajan para reducir la resistencia a la insulina; también parecen disminuir la inflamación.En varios ensayos controlados con placebo, los pacientes tratados con rosiglitazona mostraron una mejora de la memoria y de la función cognitiva.21,22,28 aunque solo se han realizado pequeños estudios de pioglitazona, este medicamento también se ha asociado con mejoras en la memoria y la cognición.,22 aunque la pioglitazona y la rosiglitazona tienen poca permeabilidad a la BBB, cada uno de estos agentes se ha documentado en el cerebro después de la administración oral.22 aunque estos medicamentos pueden desempeñar un papel en la prevención o el tratamiento de la EA, actualmente no están aprobados por la FDA para pacientes con EA, y sus posibles beneficios y riesgos deben considerarse a fondo de manera individual.
ejercicio aeróbico: quizás el método más efectivo y el más pasado por alto para reducir la resistencia a la insulina es la actividad física., La obesidad se ha asociado con un mayor riesgo de resistencia a la insulina, diabetes, demencia y EA.29,30 el ejercicio mejora la sensibilidad a la insulina y reduce los niveles periféricos de insulina.26,31 la actividad física, independientemente del peso, se asocia con un menor riesgo de EA, por lo que es la única terapia neuroprotectora comprobada.29-31 un posible mecanismo de esta neuroprotección es que el ejercicio promueve el aclaramiento de las proteínas beta-amiloides y aumenta el factor neurotrófico derivado del cerebro, un factor de crecimiento vital para la cognición y la supervivencia neuronal que se reduce en la EA.,32
papel del farmacéutico
Los farmacéuticos desempeñan un papel esencial en la educación de los pacientes sobre las nuevas terapias; también necesitan mantenerse al tanto de los nuevos descubrimientos médicos o condiciones que se presentan con frecuencia en las noticias. La diabetes tipo 3 es una de esas afecciones. Si bien queda mucho por aprender sobre este posible nuevo vínculo entre la diabetes y la cognición deteriorada, el público preguntará la opinión de sus proveedores de atención médica. Además, muchos pacientes con diabetes estarán interesados en aprender más y recurrirán a sus farmacéuticos para obtener información., Por lo tanto, los farmacéuticos deben discutir la comprensión actual de esta condición con sus pacientes y también aconsejarles sobre estrategias que pueden disminuir su riesgo, dadas sus comorbilidades individuales.
conclusión
se está llevando a cabo un estudio intensivo en un intento de caracterizar mejor la diabetes tipo 3 y desarrollar estrategias preventivas y terapéuticas. Hasta la fecha, no existen tratamientos específicos con eficacia demostrada en la prevención del deterioro cognitivo o la EA en pacientes con DM., Así, el manejo del deterioro cognitivo en pacientes con DM es idéntico al de pacientes sin DM, con atención específica al control glucémico y a los factores de riesgo cardiovascular. Este es el mismo enfoque ya esencial para prevenir las complicaciones relacionadas con la DM. Si bien algunos grupos de pacientes parecen tener un mayor riesgo de deterioro cognitivo, la mayoría de los pacientes con DM no tienen asociaciones clínicas bien definidas., Aunque no hay opciones de tratamiento específicas disponibles, el enfoque lógico actual es lograr el control glucémico y manejar prudentemente los factores de riesgo cardiovascular, como la hiperlipidemia y la hipertensión.
1. Stewart R, Liolitsa D. diabetes mellitus tipo 2, deterioro cognitivo y demencia. Diabet Med.
2. Xu WL, von Strauss E, Qiu CX, et al. La diabetes no controlada aumenta el riesgo de enfermedad de Alzheimer: un estudio de cohorte basado en la población. Diabetologia. 2009;52:1031-1039.3. Arvanitakis Z, Wilson RS, Bennett DA. Diabetes mellitus, demencia y función cognitiva en personas mayores., J Nutr Health Envejecimiento. 2006;10:287-291.
4. Jacobson AM, Musen G, Ryan CM, et al. Efecto a largo plazo de la diabetes y su tratamiento sobre la función cognitiva. N Engl J Med. 2007;356:1842–1852.
5. Whitmer RA. Diabetes tipo 2 y riesgo de deterioro cognitivo y demencia. Curr Neurol Neurosci Rep. 2007; 7: 373-380.
6. Steen e, Terry BM, Rivera EJ, et al. Deterioro de la insulina y la expresión del factor de crecimiento similar a la insulina y los mecanismos de señalización en la enfermedad de Alzheimer: ¿es diabetes tipo 3? J Alzheimer Dis. 2005;7:63-80.
7. Asociación Americana De Diabetes. Estándares de atención médica en diabetes-2010., Cuidado De La Diabetes.
8. Triplitt Cl, Reasner CA, Isley WL. Diabetes mellitus. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Principios de farmacoterapia & práctica. 7th ed. Nueva York, NY: McGraw Hill Medical; 2008: 1205-1241.
9. Nathan DM, Buse JB, Davidson MD, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Cuidado De La Diabetes.10. Hebert LE, Scherr PA, Bienias JL, et al., Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119-1122.11. Cummings J. enfermedad de Alzheimer. N Engl J Med. 2004;351:56-67.12. Slattum PW, Swerdlow RH, Hill AM. Enfermedad de Alzheimer. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Principios de farmacoterapia & práctica. 7th ed. Nueva York, NY: McGraw Hill Medical; 2008: 1051-1065.13. Craft S, Watson GS. Insulina y enfermedad neurodegenerativa: mecanismos compartidos y específicos. Lancet Neurol. 2004;3:169-178.14. de La Monte SM, Wands JR., Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimer Dis. 2005;7:45-61.15. Park CR, Seeley RJ, Craft S, Woods SC. La insulina Intracerebroventricular mejora la memoria en una tarea de evitación pasiva. Physiol Behav. 2000;68:509-514.16. Fehm HL, Perras B, Smolnik R, et al. Manipulación de las vías neuropeptidergicas en humanos: ¿un nuevo enfoque de la Neurofarmacología? Eur J Pharmacol. 2000;405:43-54.17. Zhao WQ, ALKON DL. Papel de la insulina y el receptor de insulina en el aprendizaje y la memoria., Endocrinol De Células Mol. 2001;177:125-134.18. Kopf s, Baratti C. Effects of posttraining administration of insulin on retention of a habituation response in mice: participation of a central cholinergic mechanism. Neurobiol Aprender Mem.19. Revill P, Moral MA, Prous JR. alteración de la señalización de la insulina y la patogénesis de la enfermedad de Alzheimer. Drugs Today (Barc) (En Inglés). 2006;42:785-790.20. Moroz N, Tong M, Longato L, et al. Neurodegeneración limitada tipo Alzheimer en obesidad experimental y diabetes mellitus tipo 2. J Alzheimer Dis. 2008;15:29-44.21. Haan MN., Therapy insight: diabetes mellitus tipo 2 y el riesgo de enfermedad de Alzheimer de inicio tardío. Nat Clin Pract Neurol. 2006;2:159-166.22. Landreth G, Jiang Q, Madrekar S, et al. Ppargamma agonistas como terapéutica para el tratamiento de la enfermedad de Alzheimer. Neurotherapeutics. 2008;5:481-489.23. Lyn-Cook LE, Lawton M, Tong M, et al. La ceramida hepática puede mediar la resistencia a la insulina cerebral y la neurodegeneración en la diabetes tipo 2 y la esteatohepatitis no alcohólica. J Alzheimer Dis.24. Tong M, de La Monte SM. Mecanismos de la neurodegeneración mediada por ceramida. J Alzheimer Dis.25., Reger MA, Watson GS, Green PS. La insulina Intranasal mejora la cognición y modula el beta-amiloide en la EA temprana. Neurología. 2008;70:440-448.26. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age – and obesity-related effects on memory, amyloide and inflammation (en inglés). Neurobiol Envejecimiento. 2005; 26 (suppl 1): 65-69.27. Jiang Q, Heneka M, Landreth GE. The role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in Alzheimer’s disease: therapeutic implications. Medicamentos para el SNC. 2008;22:1-14.28. Watson GS, Cholerton BA, Reger MA, et al., Conservación de la cognición en pacientes con enfermedad de Alzheimer precoz y deterioro cognitivo amnésico leve durante el tratamiento con rosiglitazona: un estudio preliminar. Am J Geriatr Psiquiatría. 2005;13:950-958.29. Luchsinger JA, Mayeux R. adiposidad y enfermedad de Alzheimer. Curr Alzheimer Res. 2007; 4: 127-134.30. Whitmer RA. La epidemiología de la adiposidad y la demencia. Curr Alzheimer Res. 2007; 4: 117-122.31. Watson GS, Craft S. the role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. Medicamentos para el SNC. 2003;17:27-45.32. Cole GM, Frautschy SA., El papel de la insulina y la señalización del factor neurotrófico en el envejecimiento cerebral y la enfermedad de Alzheimer. Exp Gerontol. 2007;42:10-21.
















Deja una respuesta